
In the Wood-Ljungdahl pathway (WLP), nature has devised one of the most elegant methods for the chemical transformation of carbon.1 The WLP is ancient, present in the last universal common ancestor to life on earth and is the only carbon fixation pathway known to produce more energy than is consumed in terms of net ATP. The WLP functions to scrub our atmosphere of the key greenhouse gas CO2, utilizing the carbon for energy and for the creation of cellular building material. This process occurs within the context of a large multi-unit protein complex made of carbon monoxide dehydrogenase (CODH) and acetyl-CoA synthase (ACS) in conjunction with a separate corrinoid iron-sulfur protein (CFeSP), which donates a methyl group to the ACS active site, resulting in the formation of acetate in the form of acetyl-CoA from the original CO2 molecule. Uniquely, this pathway can also function in both the forward and reverse directions, consuming or producing CO2 according to the needs of the organism within the context of its surrounding environment. Globally, the WLP as utilized by a myriad anaerobe, produces 1013 Kg of acetate annually, bearing the hallmarks of an ideal system off which to base biomimetic catalyst development for atmospheric CO2 capture, if only we can understand its underlying mechanism.
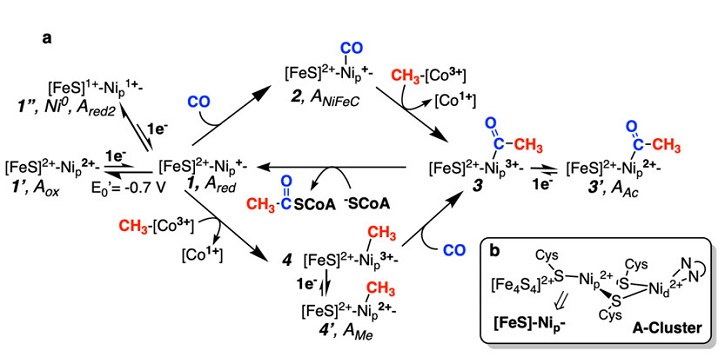
The two key enzymes of the WLP, CODH and ACS are linked as a large multimeric complex, wherein a CODH dimer is linked to two ACS units, and joined internally by a series of gas tunnels up to 140 Å1 in length which are used to shuttle CO from the CODH active site (where CO2 is converted to CO) to the active site of ACS (where CO used in acetogenesis). At the heart of CODH and ACS are unique nickel- and iron-based active sites, where acetate and acetyl-CoA are formed through a series of Ni-based organometallic intermediates (Figure 1). The active site of ACS contains six metals, four of which are irons in the form of a cubic iron-sulfur cluster, and two nickels. One nickel (the proximal Ni, or Nip) is linked to the iron-sulfur cluster by way of a bridging cysteine residue. It is anchored to a further nickel (the distal Ni, or Nid) by two additional cysteine residues. The Nid is further anchored into the peptide backbone of the ACS active site (Figure 1). In ACS, the exact mechanism of acetate synthesis from CO and CH3 has been an area of intense study for several decades, with two proposed mechanisms at the center of debate. In both mechanisms, the CO and CH3 reactants bind to the Nip, with each mechanisms positing a different oxidation state and electronic configuration for the reactant-bound Nip (Figure 1). In one mechanism, called the paramagnetic mechanism, CO and CH3 react with the Nip site to form a series of paramagnetic Ni(I) and Ni(III) intermediates, whereas in the other mechanism, the catalytically active Nip species are formally diamagnetic Ni(0) and Ni(II).
In a study led by Ragsdale (University of Michigan, Ann Arbor) and Sarangi (SSRL) et al., and in collaboration with Carnegie Mellon University and Northwestern University scientists the geometric and electronic structures of the remaining uncharacterized organometallic intermediates of ACS have been solved.
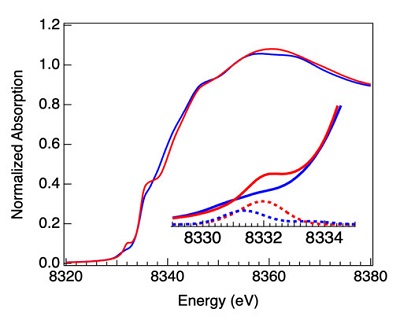
Figure 2. Ni K-edge XANES of AOx (blue) and Nip(II)-Methyl (red) ACS. The inset provides more detail of the difference in the pre-edge features, with TD-DFT simulations of the XAS spectra obtained from broken-symmetry DFT calculations shown as dotted lines.
Prior work from the Ragsdale lab has shown that that reductive activation in the presence of a substrate (CO or CH3) of the resting active site cluster is needed to jump start the cycle and together the PI’s also characterized the structure of the CO-bound intermediate through data collected at SSRL.2 In the current study, resting ACS (AOx), was reacted with a methyl donor (methyl-cob(III)inabmide, which mimics the native methyl-donor CFeSP) in the presence of reductant to form the methylated intermediate. UV-Vis experiments support a SN2 methyl transfer mechanism and suggest the formation of a paramagnetic intermediate (conversion of intermediate 1 to intermediate 4 in Figure 1a). However, EPR, ENDOR and Mössbauer analysis reveal the lack of relevant paramagnetic signal suggesting that the intermediate is diamagnetic.
Further characterization relying on high-resolution Co, Ni and Fe K-edge XAS and EXAFS conducted at SSRL’s biological beamline 7-3 showed that this methyl transfer to AOx is accompanied by a two electron reduction of the cob(III)inamide and methyl binding occurs at the Nip site -simultaneously visualizing the reduction of a Co(III) to a Co(I) species and formation of a short organometallic Ni-Methyl bond at 1.98 Å. The experimental data were combined with broken symmetry DFT calculations and TD-DFT simulations of the Ni and Fe K-pre-edges, which confirm the formation of a Nip(II)-Me group. These results indicated that a high-valent Nip(III)-Me species must be formed that gets spontaneously reduced to a Nip(II)-Me under the conditions of the study. Interestingly however, the authors observed the formation of the acetylated intermediate via incubation of the Nip(II)-methyl intermediate with CO, demonstrating that the reduced Nip(II)-methyl intermediate can be recruited back into the catalytic mechanism for acetate synthesis. The authors also characterized the Ni-acetyl intermediate using Ni and Fe K-edge XAS measurements and showed that the Ni-C organometallic bond in the Nip(II)-acetyl intermediate shrinks to 1.90 Å, thus trapping the final Ni-bound intermediate formed after C-C bond fusion.
To explain the existence of both catalytically relevant paramagnetic Ni(III) and diamagnetic Ni(II) species the authors propose a novel electrochemical (EC) coupling mechanism (see Figure 1). Under this mechanistic framework, Nip(I) (or Nip(I)-CO) reacts with methyl-Co(III) to generate Nip(III)-methyl (or Nip(III)-acetyl), which are reducible to their Nip(II) counterparts via interaction with excess reductant. These reduced intermediates are then recruited back into active catalysis through binding of the next substrate. The work presented in this study completes the description of the major intermediates in the ACS catalytic cycle. Critically, the results of this study offer a new lens through which to view the mechanism of ACS, providing a bridge between the paramagnetic and diamagnetic mechanisms and this new EC mechanism provides the framework to understand how ACS transits through diamagnetic Nip(II)-methyl and Nip(II)-acetyl intermediates (characterized in this study) while also recruiting transient Nip(I), Nip(I)-CO and Nip(III)-methyl and Ni(III)-acetyl species within the catalytic cycle.
(1) S. W. Ragsdale, "Biological Carbon Fixation by an Organometallic Pathway: Evidence Supporting the Paramagnetic Mechanism of the Nickel-Iron-Sulfur Acetyl-CoA Synthase", in Comprehensive Coordination Chemistry III; Elsevier, 2021; pp 611–633. doi: 10.1016/B978-0-08-102688-5.00082-9.
(2) M. Can, L. J. Giles, S. W. Ragsdale and R. Sarangi, "X-ray Absorption Spectroscopy Reveals an Organometallic Ni-C bond in the CO-treated Form of Acetyl-CoA Synthase", Biochemistry 56, 1248 (2017) doi: 10.1021/acs.biochem.6b00983.
M. Can, M. J. Abernathy, S. Wiley, C. Griffith, C. D. James, J. Xiong, Y. Guo, B. M. Hoffman, S. W. Ragsdale and R. Sarangi, "Characterization of Methyl- and Acetyl-Ni Intermediates in Acetyl CoA Synthase Formed during Anaerobic CO2 and CO Fixation", J. Am. Chem. Soc. 145, 13696 (2023) doi: 10.1021/jacs.3c01772.
