
Methane is the simplest organic compound with the highest energy content of any carbon-based fuel. Methane accounts for almost a quarter of U.S. energy consumption, with one-half of homes using natural gas as their heating fuel.1-2 Interestingly, more than 90% of all methane on earth is produced biogenically by methanogens which are responsible for enzymatic synthesis of 1 billion tons of methane per year.3-4 Much of the methane formed by methanogens is captured and used as an energy source by aerobic and anaerobic methanotrophic microbes.5-6 However, the increased mining of methane and industrial farming of cattle has created a mismatch between the sources and the sinks of methane, causing its atmospheric levels to double over the past two centuries. This is an environmental concern related to climate change because pound for pound, methane causes 25 times more global warming over a 100-year period than CO2.7 Thus, understanding the biosynthesis of methane is imperative from basic energy, economic and environmental perspectives.
Methane is formed by one of the few Ni containing cofactors (F430) as part of the active site of methyl coenzyme M reductase (MCR), a strictly anaerobic enzyme present in methanogenic archaea. MCR catalyzes the reaction of methyl-coenzyme M (CH3−SCoM) with coenzyme B (HSCoB) to form methane and the heterodisulfide CoMS−SCoB.8 Besides the importance of methane metabolism in our environment and global energy landscape, the basic chemistry and biology of alkane activation and formation by MCR are of fundamental interest. Because of methane’s strong sp3 C−H bonds, low solubility in both polar and nonpolar solvents, and very high ionization energy, selective activation and functionalization of the C−H bond of methane was dubbed one of the holy grail reactions in chemistry.9 MCR accomplishes this reaction, as well as methane synthesis, using a reduced Ni(I)−hydrocorphin cofactor called F430 related to porphyrin, chlorophyll, and vitamin B12. Second-sphere amino acid residues present in MCR assist in enzymatic formation of methane.10-12
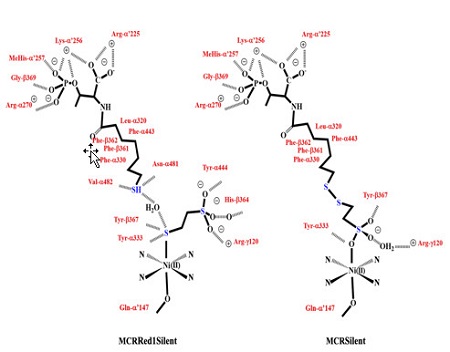
The active site of MCR has been interrogated with X-ray diffraction and the data reveal that the Ni cofactor F430 is buried at the end of a 30 Å long substrate channel. The F430 is anchored via a combination of electrostatic and H-bonding interactions between the carboxylate side-chains and the protein backbone. The four nitrogen ligands in the F430 tightly bind the Ni along the equatorial plane, while a weak distal Ni−O bond (O-α′Gln) is present in all forms of the Ni(II) enzyme as well as its active Ni(I) states.13 This ligand moves closer or away from the Ni based on the presence or absence of the upper axial ligand and may play a role in fine tuning the redox potential of the Ni center and/or provide stability during catalysis. Both substrates enter the substrate channel in a strict order with CH3−SCoM first followed by HSCoB resulting in a productive complex.14 Homolytic cleavage of the C-S bond in CH3-SCoM, is then initiated by the reduced Ni(I) form to generate a CH3· (methyl radical) that then abstracts the H from HSCoB forming methane. This is followed by S−S bond fusion and regeneration of the Ni(I) active site.15 Crystal structures on the oxidized Ni(II) form (MCRred1silent) have shown that substrate analogues HSCoM with HSCoB bind in the substrate alcove with the S of HSCoM forming a S-Ni bond to F430. This structural data has contributed to the development of a reaction mechanism that involves the formation of a Ni(I)-S bond with CH3−SCoM resulting in a methyl radical. This methyl radical will go on to generate a CoBS· radical on H abstraction. However, such binding, if catalytically relevant, leaves more than a 6 Å distance between the CoBS· and the bound S-atom of HSCoM posing a structurally “unreachable” conundrum for facile recombination to form CoMS-SCoB. Interestingly, the CoMS−SCoB binds to the Ni(II) center with its sulfotate oxygen (Figure 1) in the MCRsilent form.13 Since the published MCR mechanisms extrapolate structural details from the oxidized Ni(II) forms to the active Ni(I) form, such extrapolations are called to question in the face of the distance conundrum.
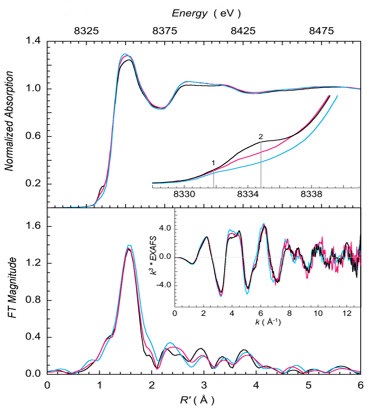
Fig 2. (top) Ni K-edge XAS data for the active Ni(I) form bound to MCRred1 showing pre-edge changes upon CoMS−SCoB binding to the Ni(I) center. (bottom) A comparison of EXAFS data reveals a unique Ni(I) hexacoordiate active site.
In a recent study the Ragsdale group at the University of Michigan in collaboration with Dr. Ritimukta Sarangi at SSRL have combined near-infrared (NIR), XAS, and EPR results to characterize the active Ni(I) state of MCR. The experimental data were combined with QM-MM and TD-DFT studies on the chemical models to successfully describe the binding details. Ni K-edge XAS and EXAFS measurements were performed at BL7-3 on the active Ni(I) state bound to CoMS−SCoB and CoMS−SCoB6, which is formed by the reaction of CH3−SCoM and the slower substrate HSCoB6 to reveal that CH3−SCoM binds to the active Ni(I) state of MCR through its sulfonate ligand, in the process forming a hexacoordinate Ni(I) complex (Figure 2).
This is remarkable, since Ni(I) complexes are usually four or five-coordinate and a six-coordinate Ni(I) is unprecedented in metalloenzyme active sites. The proposed binding mode addresses and allows the authors to provide a possible resolution to the difficult recombination of the CoBS radical with the Ni−SCoM species over a distance of 6.4 Å to generate the heterodisulfide product. The binding of CoMS−SCoB through the sulfonate also provides a viable mechanism of C−H bond activation going in the reverse direction in the situation where the disulfide bond is unable to get closer to the Ni(I) due to the anchoring of the substrate at the top of the channel by electrostatic residues.13 This revised mechanism obviates the need for substantial active site rearrangements that may involve large thermodynamic and kinetic penalties and provides a unifying solution based on long range electron transfer from the Ni(I) to the substrate. The study was highlighted in JACS Spotlight as “groundbreaking clarification of Ni(I)–sulfonate binding” (DOI: 10.1021/jacs.1c03484).
1. E. F. DeLong, Nature 407, 577 (2000)
2. S. K. Atreya, P. R. Mahaffy, and A. Wong, "Methane and Related Trace Species on Mars: Origin, Loss, Implications for Life, and Habitability", Planetary Space Sci. 55, 358 (2007)
3. D. A. Lashoff and D. Ahuja, "Relative Contributions of Greenhouse Gas Emissions to Global Warming", Nature 344, 213 (1990)
4. U. Ermler, W. Grabarse, S. Shima, M. Goubeaud, and R. K. Thauer, "Crystal Structure of Methyl-coenzyme M Reductase: The Key Enzyme of Biological Methane Formation", Science 278, 1457 (1997)
5. T. Wongnate and S. W. Ragsdale, "The Reaction Mechanism of Methyl-coenzyme M Reductase: How an Enzyme Enforces Strict Binding Order", J. Biol. Chem. 290, 9322 (2015)
6. T. Wongnate, D. Sliwa, B. Ginovska, D. Smith, M. W. Wolf, N. Lehnert, S. Raugei, and S. W. Ragsdale, "The Radical Mechanism of Biological Methane Synthesis by Methyl-coenzyme M Reductase", Science 352, 953 (2016)
A. Patwardhan, R. Sarangi, B. Ginovska, S. Raugei and S. W. Ragsdale, "Nickel–Sulfonate Mode of Substrate Binding for Forward and Reverse Reactions of Methyl-SCoM Reductase Suggest a Radical Mechanism Involving Long-range Electron Transfer", J. Am. Chem. Soc. 143, 5481 (2021) doi: 10.1021/jacs.1c01086
