
Voltage-gated sodium (Nav) channels are primarily responsible for initiating action potentials in nerves, muscles, and heart1,2. Dysfunction of these channels leads to diseases including epilepsy, arrhythmia, and chronic pain. Humans have nine channel subtypes, Nav1.1 to 1.9, and each has distinct expression patterns, functional properties, and physiological roles. Nav1.7 is highly expressed in peripheral sensory neurons and plays a critical role in pain sensation. Mutations in Nav1.7 have been characterized in many pain syndromes, including both extreme pain disorder and indifference to pain. Understanding the detailed structure of Nav1.7 in complex with toxin modulators might facilitate drug discovery for this promising chronic pain target. However, efforts to obtain such structures have been hindered by technical challenges in recombinant protein production and high-resolution structural analysis by approaches such as x-ray crystallography.
Spider venom contains toxins that modulate Nav channels by blocking the central ion conducting pore or by modulating channel opening by binding at allosteric sites within the voltage sensing domains (VSDs). Toxins, called gating modifier toxins (GMTs), use the latter mechanism, and have been used to probe Nav channels’ complex gating properties. GMTs can exhibit different functional impacts on channel gating depending upon the VSD that they bind. Protoxin-II (ProTx2) from the Peruvian green velvet tarantula is a selective antagonist of the human Nav1.7 channel and it was previously thought to trap VSD2 in a deactivated state to stabilize a closed state of channel3,4.
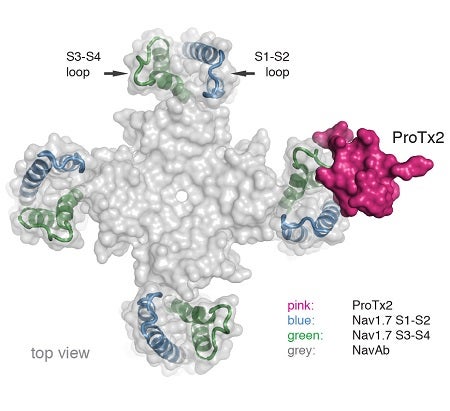
To pursue structural studies, researchers from Genentech created a chimeric channel in which the VSD of the Nav channel from the bacterium Arcobacter butzleri (NavAb) was replaced with the VSD2 from human Nav1.7. X-ray diffraction experiments were performed at three beam lines, including Beam Line 12-2 at SSRL and CXI at LCLS, to determine the crystal structure of ProTx2 bound to the VSD2-NavAb chimeric channel (Figure).
The crystal structure obtained by the researchers revealed that ProTx2 targets the S3-S4 loop of VSD2 through an electrostatic interaction. Unexpectedly, ProTx2 was found to bind VSD2 in an activated state, and functional studies on the Nav1.7 channel confirmed that ProTx2 appears to remain bound during channel opening and closing events. The researchers further performed cryo-EM analysis. Cryo-EM studies revealed a reconstruction where ProTx2 was bound to VSD2 in the deactivated state, where the S4 helix had translated 10 Å downward, and pushed the S4–S5 linker inward to close the S6 activation gate. The cryo-EM result also validated the structural model derived from high-resolution crystallographic analysis.
The x-ray crystal structure finally helps to explain the inhibitory action of ProTx2 on Nav1.7, but also shows how inhibitory GMTs like ProTx2 can bind to the resting VSD with extremely high affinity and also to the activated VSD with still relatively high affinity. Importantly, the structure also explains the high selectivity of ProTx2 for Nav1.7. Overall, this study may help researcher design new modulators to bind VSD2 during Nav channel gating to silence hyperexcitability during epileptic seizures or chronic pain flare ups.
In summary, this study revealed unprecedented insights into voltage sensing and electromechanical coupling in Nav channels and may provide a potential path for designing the next generation of Nav channel antagonists that are selective for specific channel subtypes.
1. W. A. Catterall, “Molecular Properties of Voltage-sensitive Sodium Channels”, Annu. Rev. Biochem. 55, 953 (1986).
2. J. Payandeh and D.H. Hackos, “Selective Ligands and Drug Discovery Targeting the Voltage-Gated Sodium Channel Nav1.7”, Handb. Exp. Pharmacol., 246, 271 (2018).
3. M. Flinspach, et al., “Insensitivity to Pain Induced by a Potent Selective Closed-state Nav1.7 Inhibitor”, Sci. Rep., 7, 39662 (2017).
4. F. Bosmans, M. F. Martin-Eauclaire, K. J. Swartz, “Deconstructing Voltage Sensor Function and Pharmacology in Sodium Channels”, Nature, 456, 202 (2008).
H. Xu, T. Li, A. Rohou, C. P. Arthur, F. Tzakoniati, E. Wong, A. Estevez, C. Kugel, Y. Franke, J. Chen, C. Ciferri, D. H. Hackos, C. M. Koth and J. Payandeh, "Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin", Cell 176, 702 (2019) doi: 10.1016/j.cell.2018.12.018
