

Mammalian hosts defend against invading pathogens via the import of toxic concentrations of copper into the phagolysosome. To combat this host-defense strategy, gram negative pathogens respond via sophisticated copper export systems which are able to neutralize the copper onslaught2. Chemical mechanisms of metal exchange between protein components of metal exporters are thus important factors in understanding pathogenic virulence and are believed to occur via formation of intermediates in which the metal is coordinated by ligands derived from each partner. However, since these ligand sets are often similar (or even identical), following the kinetics of transfer has been challenging, and has required the development of sophisticated spectroscopic approaches.
The CusCBAF exporter of E. coli is an example of a molecular exporter that combats copper-mediated host defense3 (Figure 1). In earlier work published in PNAS4 and reported as an SSRL Highlight we laid the groundwork for detailed mechanistic studies by using selenomethionine (SeM) labeling and multi-edge EXAFS to show that CusB activates CusA to accept Cu(I) ions from the CusF metallochaperone in a reversible reaction. First CusF senses the periplasmic copper load at its Met2HisTryp metallosite, then transfers Cu(I) to the Met3 binding site on CusB, allowing the latter to bind and activate the CusA exporter to accept copper also from CusF. As periplasmic copper levels fall, copper is back-transferred from B to F, switching off the pump.
Following on from these studies we have now successfully captured and characterized a shared ligand transfer intermediate between CusF and CusB containing two Met ligands from CusB and one Met from CusF. This chemistry provides the first direct experimental evidence for the paradigm of shared ligand transfer intermediates in chaperone-target interactions, and provides experimental validation of earlier in silico predictions of the intermediate5. The work carried out on SSRL Beam Lines 7-3 and 9-3 was made possible by the development of a robust technique for interrogating the kinetics of metal transfer via SeM labeling the methionine ligands of one member of the chaperone-target pair, and monitoring metal transfer in real time (rapid-freeze quench) using the intensity of the Se-Cu signal in the Fourier transform of the Se EXAFS (Fig. 2). In a reaction between Cu(I)-loaded CusF and SeM-labeled apo-CusB, the intensity of this signal was found to build to a level characteristic of the shared ligand intermediate in ~50 ms and then slowly (~1 s) convert to products at 25°C. The intermediate could be stabilized at 10°C, and its coordinate structure identified by sequential studies of the Se and Cu EXAFS. Parallel studies on the rate of Cu(I)-mediated quenching of the W44 trypophan fluorescence of CusF also indicated a 2-step process that could be interpreted as (i) formation of a protein-protein complex (fast) with copper sharing ligands from CusB and CusF, and (ii) resolution of this complex to products (slow).
The chemical speciation of the shared ligand intermediate stabilized at 10°C was dependent on which of the two partners was labeled with SeM, suggesting that the thermodynamics of intermediate formation was influenced by subtle differences in the bond energies of the metallosites. This is consistent with the premise that metal transfer is driven by a shallow thermodynamic gradient, and that selectivity arises from molecular recognition between chaperone and target pairs. With the label at the Met3 site of CusB, the intermediate was comprised of one Met from CusF and two Mets from CusB; however, when the label was located at the Met2His site of CusF, a different intermediate (Fig. 2) was stabilized involving a Met and a His from CusF and one Met from CusB. We postulated that both intermediates may exist along the reaction coordinate, and that the one that is stabilized depends on the relative stabilities of the metal-ligand bonds. The rate constants estimated for protein-protein complex formation indicated a diffusion controlled process suggestive of a driving force partially assisted by the formation of the shared ligand complex. Thus, the resulting chemical mechanism represents a careful balance between the energetics of molecular recognition, shared-ligand intermediate stability and the rate of its decay to products.
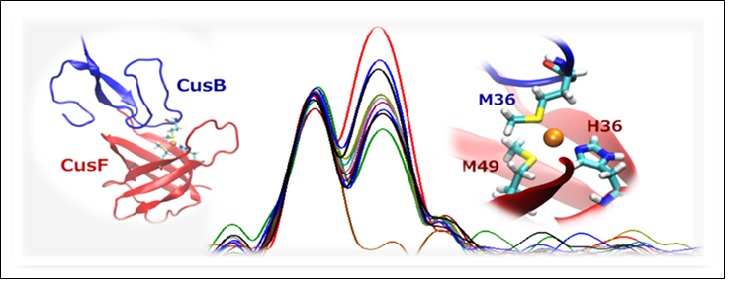
Figure 2. Middle: Se EXAFS Fourier transforms at increasing time points for the reaction of SeM-labeled apo-CusF with unlabeled Cu(I)-loaded CusB. Left and right: in silico models of the proposed protein-protein interface and shared-ligand intermediate.
- M. N. Ucisik, D. K. Chakravorty and K. M. Merz, Jr., "Structure and Dynamics of the N-terminal Domain of the Cu(I) Binding Protein CusB", Biochemistry 52, 6911 (2013)
- J. J. Braymer and D. P. Giedroc, "Recent Developments in Copper and Zinc Homeostasis in Bacterial Pathogens", Curr. Opin. Chem. Biol. 19C, 59 (2014)
- T. D. Mealman, N. J. Blackburn and M. M. McEvoy, "Metal Export by CusCFBA, the Periplasmic Cu(I)/Ag(I) Transport System of Escherichia coli", Curr. Top. Membr. 69, 163 (2012)
- K. N. Chacón, T. D. Mealman, M. M. McEvoy and N. J. Blackburn, "Tracking Metal Ions through a Cu/Ag Efflux Pump Assigns the Functional Roles of the Periplasmic Proteins, Proc. Natl. Acad. Sci. USA 111, 15373 (2014)
- M. N. Ucisik, D. K. Chakravorty and K. M. Merz, Jr., "Models for the Metal Transfer Complex of the N-terminal Region of CusB and CusF", Biochemistry 54, 4226 (2015)
K. N. Chacón, J. Perkins, Z. Mathe, K. Alwan, E. N. Ho, M. N. Ucisik, K. M. Merz and N. J. Blackburn, "Trapping Intermediates in Metal Transfer Reactions of the CusCBAF Export Pump of Escherichia coli", Commun. Biol. 1, 192 (2018) doi: 10.1038/s42003-018-0181-9
