
Copper serves as a redox center in metalloproteins, often cycling between the +1 and +2 oxidation states. Oxidases, such as petidylglycine monooxygenase (PHM), bind oxygen at Cu(I) sites giving rise to "oxo" reactive intermediates capable of oxidizing organic substrates. To modulate the redox properties of the copper center, the coordination sphere at the metal consists of a mixture of cupriphilic histidines (His) and cuprophilic methionie (Met) residues. On the other hand, transport proteins favor Met ligation over His which stabilizes the metal center in the Cu(I) form prevalent in the reducing environment of the cell. A major question relevant to copper biochemistry is how the ligand environment influences reactivity of the site towards either oxygen-based catalysis or Cu(I)-based transport. Answering this fundamental question is made more challenging by the paucity of spectroscopic approaches amenable for studying metals such as Cu(I) in which the d-shell is full.
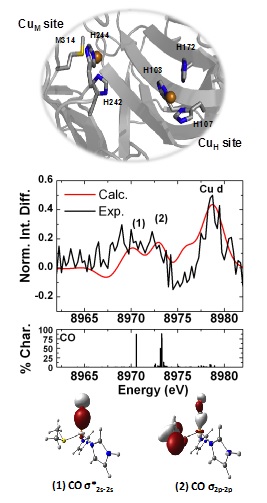
Figure 1. (top) PHM Cu binding sites; (middle) Overlay of experimental and calculated VtC XES difference spectra of PHM-Co and PHM with the percent Co character breakdown for individual transitions; (bottom) Molecular orbitals corresponding to features of interest (isovalue 0.05) .
Given the fact that Cu(I) centers in proteins are often comprised of mixed histidine-methionine ligand sets, we set out to develop new approaches for understanding their electronic contributions to reactivity. In recent years valence-to-core x-ray emission spectroscopy (VtC XES) has become a powerful technique to characterize the ligand environment at the metal center. Non-resonant Kb VtC involves excitation of core electrons and detection of emissions from filled valence orbitals generally dominated by ligand orbitals having a s interaction with the metal. Emission energies are resolved by employing a spectrometer such as the one at SSRL Beam Line 6-2 consisting of a multi-crystal analyzer in Rowland geometry. VtC XES is particularly adept at characterizing metal ligand interactions, especially in the case of small molecules, being sensitive to not only speciation but binding mode as well.2
The ligand environment for a series of Cu(I)-metalloproteins with a systematic variation in Met-His ligation was investigated. These included the metal transporters CusB (Met3) and CusF (Met2His) along with PHM variants having only the CuM (Met1His2) site or CuH (His3) site. Although previous Cu K-edge XAS studies on the series had confirmed the presence of three coordinate Cu(I) sites,1 the lack of pre-edge features and the inability to differentiate between nitrogen, oxygen or carbon donors limits the quantitative information that can be obtained. The latter is particularly important as the CuM site is known to bind small molecules such as H2O, O2 or CO to form a four coordinate metal center. VtC XES was employed to characterize the Cu(I) ligand environment in each of the mixed ligand complexes to determine the impact of changing the ratio of His to Met ligands, and the impact of binding the oxygen surrogate CO at the CuM site of wild type PHM (having both CuM and CuH sites present - Figure 1 (top).
The study shows that in addition to being sensitive to the nature of ligand speciation and ligand binding mode, features from the Cu 3d orbitals are also observed and are most intense at the Met3 limit. Correlation of the spectroscopic data with theoretical calculations and in silico model studies showed that Cu 3d emissions gain intensity from ligand-mediated Cu p-d mixing, through a configuration interaction mechanism and are dependent on the metal-ligand orbital overlap and local metal geometry. With this information in hand, the applicability of VtC XES for mechanistic studies was investigated by exploring the changes in the PHM spectrum upon CO binding to the CuM site. To highlight the changes, the difference spectrum between PHM-CO (wild type PHM with CO bound) and PHM (constructed from the average spectra of the CuM and CuH variants) is shown in Figure 1. Two features at ~8970.0 eV (1) and ~8972.5 eV (2) are observed to grow in the presence of CO. When correlated with theoretical calculations these features are seen to arise from the C-O s s*2s-2s and C-O s2p-2p orbitals consistent with previous reports.3 Furthermore the covalent interaction between the Cu(I) center and CO increases p-d mixing into the Cu d orbitals resulting in an increase in the intensity of the corresponding feature at 8978.3 eV. In summary the study offers the promise of using VtC in conjunction with x-ray absorption spectroscopy to offer a more complete picture of the electronic structure and ligand environment at the metal center, particularly relevant in the case of electron-rich metal centers such as those having filled d10 shells. Furthermore, VtC is sensitive to small molecule binding even in the presence of discrete metal sites with varying coordination environment, suggesting it would be an appropriate method for future mechanistic studies.
[1] (a) S. Chauhan, C. D. Kline, M. Mayfield and N. J. Blackburn, Biochemistry 53, 1609 (2014); (b) I. R. Loftin, S. Franke, N. J. Blackburn and M. M. McEvoy, Protein Sci. 16, 2287 (2007); (c) I. Bagai, W. Liu, C. Rensing, N. J. Blackburn and M. M. McEvoy, J. Biol. Chem. 282, 35695 (2007).
[2] J. A. Rees, V. Martin-Diaconescu, A. J. Kovacs and S. DeBeer, Inorg. Chem. 54, 6410 (2015).
[3] M. U. Delgado-Jaime, S. DeBeer and M. Bauer, Chem. Eur. J. 19, 15888 (2013).
V. Martin-Diaconescu, K. N. Chacόn, M. U. Delgado-Jaime, D. Sokaras, T.-C. Weng, S. DeBeer and N. J. Blackburn, "Kβ Valence to Core X-ray Emission Studies of Cu(I) Binding Proteins with Mixed Methionine – Histidine Coordination. Relevance to the Reactivity of the M- and H-sites of Peptidylglycine Monooxygenase", Inorg. Chem. 55, 3431 (2016), doi: 10.1021/acs.inorgchem.5b02842.
