
Proteins interact with one another by adopting complementary shapes with complementary chemical groups. It should therefore be possible to create new biological functions by designing proteins with novel nanostructures that can interact.
Until recently, a detailed biophysical understanding of how proteins interact was elusive. Many natural protein interfaces had been modified and studied, and a few attempts had been made at creating protein interfaces de novo. But to succeed in robust protein interface design, more needed to be understood.
In recent work published in Nature, computational biologists at the University of Washington set out to discover the principles of protein interaction and use them to create novel protein interfaces. Using Rosetta-based design, the team created molecules that can target important proteins in the body, such as the insulin receptor, as well as proteins on the surface of viruses. This solves a long-standing challenge in drug development and may lead to new treatments for cancer, diabetes, infection, inflammation, and beyond.
This work combined recent advances in computational protein design to arrive at a strategy for creating new proteins that bind molecular targets in a manner similar to antibodies. The resulting software can scan a target molecule, identify potential binding sites, generate proteins targeting those sites, and then screen from millions of candidate binding proteins to identify those most likely to function. In total, the team produced over half a million candidate binding proteins. Data collected on this large pool of candidate binding proteins was used to improve the overall method.
Initially, zero success was observed across 15,000 attempts to computationally design proteins that could bind the influenza hemagglutinin H3 protein. Using human intuition as a guide, additional designs were created, and a few were found to bind to the targets IL-7 and TrkA. With successful initial designs and their site saturation mutagenesis libraries to examine, a few principles could be observed: Rosetta, the protein design software, appeared to model well the important hydrophobic interface interactions, but side-chain packing and overall interface hydrophobicity mattered more than the software accounted for. These observations allowed for the software to be improved, allowing the team to produce additional binding proteins for 14 different targets, including the SARS-CoV-2 spike protein.
Cross-reactivity studies among all the binder-target pairs show that the proteins were specific for their intended target, and competition studies suggested that the binders adhered to the right spot on the target. But the team sought further confirmation that their designed proteins were working as intended.
Crystallography experiments carried out at SSRL BL12-2 showed that the TrKA receptor binder design matched the structure (Fig.1 C & D). In total, structures for five of the designed protein structures were determined based on data collected from SSRL, ESRF and APS, revealing that all five bound to their targets as intended. For all five crystallized structures, the binding mode was within 2 Å RMSD of the design model.
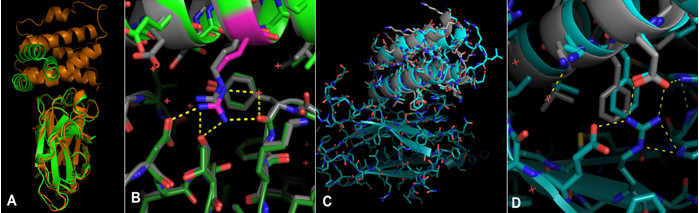
With principles for designing protein interfaces in hand, the door is now open to creating innovative new therapeutics, diagnostics, and tools. The SARS-CoV-2 binder neutralizes the virus, and the cell surface receptor binders can be used to target specific cell types (unpublished data) or to block native pathways.
The research team included scientists from the University of Washington School of Medicine, Yale University School of Medicine, Stanford University School of Medicine, Ghent University, The Scripps Research Institute, and the National Cancer Institute, among other institutions.
L. Cao, B. Coventry, I. Goreshnik, B. Huang, J. S. Park, K. M. Jude, I. Markovic, R. U. Kadam, K. H. G. Verschueren, K. Verstraete, S. T. R. Walsh, N. Bennett, A. Phal, A. Yang, L. Kozodoy, M. DeWitt, L. Picton, L. Miller, E.-M. Strauch, N. D. DeBouver, A. Pires, A. K. Bera, S. Halabiya, B. Hammerson, W. Yang, S. Bernard, L. Stewart, I. A. Wilson, H. Ruohola-Baker, J. Schlessinger, S. Lee, S. N. Savvides, K. C. Garcia and D. Baker, "Design of Protein Binding Proteins from Target Structure Alone", Nature 605, 551 (2022) doi: 10.1038/s41586-022-04654-9
