- Home
- Research program
- Research highlights
- Exported organic carbon promotes reducing conditions and redox cycling in oxic aquifers
- Soil organic matter controls Pb release during redox cycles in floodplain soils
- Spatial and Compositional Heterogeneities Control Zn Retention Mechanisms in a Simulated Aquifer
- Calcium-Uranyl-Carbonato Species Kinetically Limit U(VI) Reduction by Fe(II) and Result in U(V)-bearing Ferrihydrite
- Diverse Ammonia-Oxidizing Archaea Dominate Subsurface Nitrifying Communities in Semi-Arid Floodplains
- A Simplified Way to Predict the Function of Microbial Communities
- Complexation Organic Matter Controls Uranium Mobility Anoxic Sediments
- FES-Nanoclusters can mobilize Fe and S from sediment to the groundwater
- Hexavalent uranium storage mechanisms in wet-dry cycled sediments at contaminated DOE sites in the Western U.S.
- Redox-Interfaces can Produce Toxic Arsenic Levels Groundwater...
- Sorption to Organic Matter Controls Uranium Mobility
- Thermodynamic preservation of carbon in anoxic environments
- Iron and sulfur cycling in NRZs controlled by sediment textural and hydrology
- A regional model for uranium redox and mobility
- Long-Term in Situ Oxidation of Biogenic Uraninite in an Alluvial Aquifer: Impact of Dissolved Oxygen and Calcium
- U Release from NRZ sediments is inhibited by Transport and Geochemistry
- Team
- Previous research
- Publications





 The science.
The science.
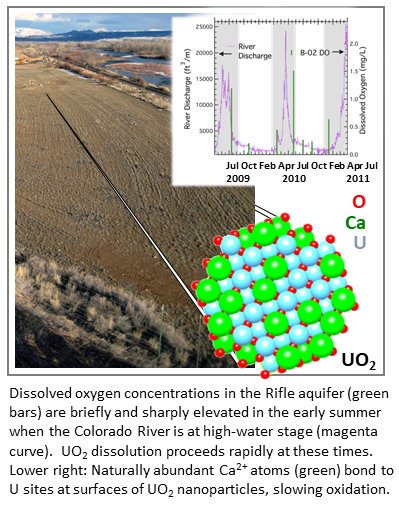
Oxidative dissolution of U(IV) to U(VI) controls the release of uranium from reduced sediments to groundwater. In this study, uraninite (UO2) was used as a proxy for all forms of subsurface U(IV) in order to study oxidative dissolution of U(IV) under bona fide aquifer conditions. Uraninite and non-crystalline forms of U(IV) occur at elevated concentrations in naturally reduced organic-rich sediments within the upper Colorado River Basin (CRB) and in sediments that have been treated with stimulated bioreduction to remediate uranium in groundwater. Prior work shows that uraninite oxidative dissolution is profoundly influenced by the concentrations of dissolved oxygen (DO), which accelerates oxidative dissolution, and Ca2+, which inhibits oxidative dissolution. In complex and heterogeneous aquifers, DO varies laterally and vertically. Moreover, large increases in DO concentrations occur during the spring/summer meltwater runoff at floodplains throughout the CRB. At the Rifle, CO, DOE field research site, the location of field-scale in situ bioremediation studies using acetate as an exogenous electron donor, average DO varies between a low of ca 0.2 mg/L during fall and winter, and a high of ca 1.5 mg/L during spring/summer meltwater runoff. This variability challenges efforts to model uranium behavior and leads to the question; how do uraninite oxidative dissolution rates vary across space and time under these varied aquifer conditions? Knowledge of the answer to this question is needed to predict subsurface uranium behavior over the long term and to optimize the selection and performance of remediation technologies.
The objectives of this study were (i) to assess the impact of variable dissolved oxygen conditions present in the field on uraninite oxidation mechanisms and cumulative loss rates and (ii) to evaluate the structural basis by which Ca2+ (and by extension, other alkaline earths such as Mg2+) associates with uraninite under field conditions. To achieve this goal, biogenic uraninite was deployed into groundwater in wells over a 22 month interval at the Rifle site, and subsequently retrieved for analysis of uranium content, oxidation state, and local molecular-scale structure around Ca and U.
Impact. Oxidation of subsurface uraninite in the Rifle aquifer occurs almost exclusively during “hot moments” when dissolved oxygen is seasonally abundant in groundwater during meltwater runoff. However, under low DO conditions, i.e., when DO concentrations remain below ca 0.6 mg/L, uraninite can be stable over decadal time scales. These results highlight the profound importance of summer meltwater as a driver for biogeochemical contaminant cycling in CRB floodplains. Large inventories of uranium reside in organic-enriched sulfidic sediments present at the Rifle site and other contaminated aquifers across the upper CRB. Seasonal meltwater conditions are expected to drive strong pulses of uranium oxidation in these sediments, leading to mobilization of uranium to the aquifer, contributing to the persistent uranium plumes.
The present results suggest that strong bonding of calcium at uranium structural sites at uraninite surfaces is a root cause for the reported retardation of uraninite oxidative dissolution rates in the presence of Ca2+. Mg2+ is also relatively abundant in Rifle groundwater and was associated with uraninite after reaction in the aquifer. The ionic radius of Mg2+ (0.89 Angstroms) is reasonably matched to that of U4+. We expect that Mg2+ also bonds tightly to uraninite surfaces, further reducing the reactivity of the underlying uraninite.
Summary. Uraninite was reacted through three meltwater seasons. Uraninite loss was rapid when DO concentrations were > 0.6 mg/L. In contrast, no measureable uraninite loss occurred in wells for which DO was < 0.6 mg/L. This result illustrates the sensitivity of oxidative dissolution rates to DO concentration.
Chemical digestions showed that Ca2+ was present at high concentrations, comprising 25% of all metals present. Subsequently, high-resolution transmission electron microscopy, x-ray absorption spectroscopy, and x-ray diffraction were performed to determine the molecular structure around uranium and calcium and to detect the presence of non-uraninite phases. These measurements indicated that Ca2+ was bound to uraninite surfaces at structural uranium sites. Uraninite surfaces were found to be free of calcium carbonate precipitates and U(VI) solid products.
Juan S. Lezama-Pacheco†, José M. Cerrato§, Harish Veeramani‡, Daniel S. Alessi‡, Elena Suvorova‡, Rizlan Bernier-Latmani‡, Daniel E. Giammar§, Philip E. Long∥, Kenneth H. Williams∥, and John R. Bargar*†
Environ. Sci. Technol. 2015, 49, 12, 7340–7347, May 22, 2015
https://doi.org/10.1021/acs.est.5b00949
