
Role of Specific Protein Mutations in Causing Human
Disease Revealed
summary written by Brad Plummer, SLAC
Communication Office
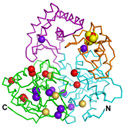
Scientists are one step closer to understanding a piece of the machinery involved in DNA transcription and repair, thanks to work done in part at the SSRL macromolecular crystallography Beam Line 11-1. The team, led by The Scripps Research Institute researcher John Tainer, and colleagues worked out the structure of an important enzyme call XPD, a member of the helicase family of enzymes, found in all living organisms. The results were published in the May 2008 edition of the journal Cell.
In eukaryotes, XPD is responsible for unwinding double-stranded DNA molecules during transcription or repair. Because it plays such a key role in the DNA transcription and repair pathways, determining the structure of XPD is crucial to understanding how healthy cells operate and how defects lead to disease. Mutations in the XPD protein are associated with cancer and premature aging (progeria).
Human XPD has proved an elusive target for crystallographic studies because it
is difficult to synthesize in sufficient quantities. Working with an XPD
homolog from the single-celled Sulfolobus acidocaldarius, the team confirmed
evidence from earlier experiments that suggested one of the four domains of the
XPD structure contains an iron-sulfur containing cluster, and provided for the
first time the structural basis to explain the mutations for three
distinguished inherited diseases.
To learn more about this research see the full Scientific Highlight
L. Fan, J.O. Fuss, Q.J. Cheng, A.S. Arvai, M. Hammel, V.A. Roberts, P.K. Cooper and J.A. Tainer, "XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations", Cell 133 (2008), pp. 789-800
