
 | |
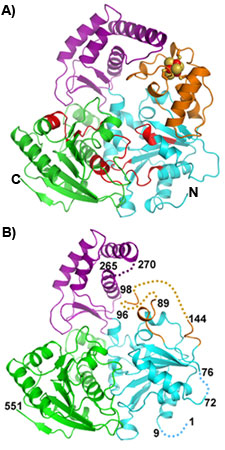
| Figure 1. Ribbon diagram of crystal structures of SaXPD (A) and apo SaXPD (B). SaXPD consists of four domains: two helicase domains (cyan and green), a 4FeS domain (orange) containing a 4Fe-4S cluster (ball-sticks), and an Arch domain (purple). Seven conserved helicase motifs are highlighted in red. Disordered regions in the apoSaXPD structure are indicated by dashed lines. | |
Using data collected at SSRL, Fan and coworkers led by John Tainer determined the crystal structure of an XPD homolog from Sulfolobus acidocaldarius (SaXPD). SaXPD is a smaller helicase than human XPD, but contains all the conserved helicase motifs and amino acid residues corresponding to 22 out of 26 disease mutations in human XPD. The crystal structure (Fig1A) reveals four domains including two motor domains (helicase domain I and II), a unique FeS domain containing a 4Fe-4S cluster with sensitivity to oxygen, and a novel Arch domain. The apo SaXPD (Fig1B) crystal structure (data collected at ALS), in which the FeS cluster was removed by soaking the crystals with oxidizing agent, indicates that the 4Fe-4S cluster is essential for the proper folding of the FeS domain and possibly plays a role in coordination of domain movements during ATP hydrolysis and DNA translocation by XPD. When mapped into the SaXPD crystal structure, the amino acid residues corresponding to 22 human diseases mutations can be distinguished into three groups matching the three different diseases (Fig2). Amino acid residues of XP mutations reside either near the ATP-binding
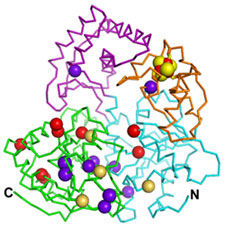 | |
| Figure 2. Distribution of amino acid residues corresponding to human XPD disease mutations. XP mutations are represented by red spheres, XP/CS mutations by greenish yellow spheres, and TTD mutations by dark purple spheres. |
Primary Citation
References
S.C. Wolski, J. Kuper, P. Hanzelmann, J.J. Truglio, D.L. Croteau, B. Van Houten
and C. Kisker, Crystal structure of the FeS cluster-containing nucleotide
excision repair helicase XPD, PLoS Biol. 6 (2008), p. e149.
J. Rudolf, V. Makrantoni, W.J. Ingledew, M.J. Stark and M.F. White, The DNA
repair helicases XPD and FancJ have essential iron-sulfur domains,
Mol. Cell 23 (2006), pp. 801-808.
L. Fan, J.O. Fuss, Q.J. Cheng, A.S. Arvai, M. Hammel, V.A. Roberts, P.K. Cooper
and J.A. Tainer, XPD helicase structures and activities: insights into the
cancer and aging phenotypes from XPD mutations, Cell 133
(2008), pp. 789-800
H. Liu, J. Rudolf, K.A. Johnson, S.A. McMahon, M. Oke, L. Carter, A.M.
McRobbie, S.E. Brown, J.H. Naismith and M.F. White, Structure of the DNA repair
helicase XPD, Cell 133 (2008), pp. 801-812.
SSRL is supported by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.