
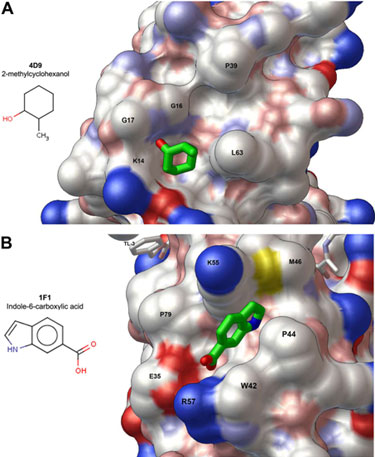
Figure 1.
Surface rendering of the HIV protease structure showing solvent-exposed clefts
on the protein surface into which the fragments bind. (A) The exo site binds
2-methylcyclohexanol, and (B) the outside/top of the flap binds
indole-6-carboxylic acid. The exo site is a pre-existing feature of the
protein fold while the outside/top of the flap rearranges to accommodate
fragment binding. These results provide a basis to develop larger, higher
affinity inhibitors specific for each site. C, N, O, and S atoms are colored
white, blue, red, and yellow, respectively.
HIV protease, with a tight binding inhibitor bound in the active site, was
co-crystallized in the presence of small molecule drug fragments. Altogether,
400 fragments were screened, over 800 crystals were evaluated, and 378 data
sets were collected to 2.3-1.3 Å resolution using the robotic sample
automounter system available at SSRL beam lines. Analysis of the data in
collaboration with SSRL staff revealed that fragment binding within each
surface site induces a distinct conformation of the protease, leading to
appearance of different crystal forms. In the shallow cleft termed the 'exo
site' [2, 3] the fragment
2-methylcyclohexanol binds adjacent to the
Gly16Gly17Gln18 loop where the amide
of Gly17 is a specific hydrogen bond donor, and hydrophobic contacts
occur with the side chains of Lys14 and Leu63 (Fig. 1A).
In a hydrophobic pocket on the outside surface of one protease flap, another
fragment, indole-6-carboxylic acid, binds via hydrophobic contacts with
Trp42, Pro44, Met46, and Lys55, a
hydrogen bond with Val56, and a salt-bridge with Arg57
(Fig. 1B). A similar fragment, 2-acetyl-benzothiophene, also binds at this
site. These results could not have been obtained without the high-throughput
capability of SSRL beam lines.
This study is the first in which fragments were screened against an
inhibitor-bound drug target. The results show that binding sites exist in HIV
protease outside the active site and establish a starting point for developing
larger, higher affinity molecules able to bind and stabilize the closed,
inhibited enzyme. By exploiting an allosteric mechanism such molecules could
act in synergy with FDA-approved inhibitors to restore potency against
multi-drug-resistant HIV mutants.
Primary Citation
Perryman, A. L., Zhang, Q., Soutter, H. H., Rosenfeld, R., McRee, D., Olson, A.
J., J. E. Elder, J. E. & Stout, C. D. (2010) Fragment-based Screen against HIV
Protease. Chem. Biol. Drug Des. 75: 257-268.
References
A team of scientists at The Scripps Research Institute using SSRL resources
have applied fragment-based crystallographic screening to HIV protease and
discovered two novel binding sites on the surface of the protein. These sites
can now be targeted to develop larger, higher affinity drug-like molecules.
HIV protease is an essential viral enzyme and important drug target in the
fight against AIDS. However, multi-drug-resistant mutations, which severely
compromise the potency of protease inhibitors, keep appearing at
ever-increasing frequencies [1]. Most mutations that cause drug resistance are
found within the active site, in the hollow center of the enzyme, which is
guarded by two highly mobile flaps. In contrast, the 'fragments', which are
molecules smaller than typical drugs, bind in pockets and clefts on the
protease surface and not in the active site. Computer simulations show that
one of these clefts must alter its shape when the flaps open and close, as
required for catalytic activity during the viral life cycle [2]. Moreover,
multi-drug-resistant mutants appear to require a greater range of freedom
within this surface binding site [3]. Consequently, larger molecules, designed
to bind in these surface sites, should complement active site specific drugs
and work to suppress the evolution of resistance by restraining the necessary
range of motion in HIV protease. The fragment screening results provide the
first key step in this novel approach to drug discovery.
SSRL is supported by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.