
Methyl-coenzyme M reductase (MCR) from methanogenic archaea
catalyzes the terminal step in biological methane synthesis. Using coenzyme B
(CoBSH) as the two-electron donor, MCR reduces methyl-coenzyme M (methyl-SCoM)
to form methane and the heterodisulfide product, CoBS-SCoM. MCR contains an
essential redox active nickel tetrapyrrolic cofactor called coenzyme
F430 at its active site, which is active in the reduced Ni(I) state
(MCRred1). All of the biologically generated methane,
amounting to 1 billion tons per annum globally, is formed by MCR. Furthermore,
recent evidence indicates that anaerobic methane oxidation is also catalyzed by
MCR and occurs by a reversal of the methane synthesis reaction. Methane is a
potent greenhouse gas, trapping 20 times more heat than CO2. In
addition, methane is also an important and clean fuel as it produced the least
amount of CO2 per unit of heat released. Thus, it is critically
important to understand the mechanism of formation of the smallest hydrocarbon
in nature.
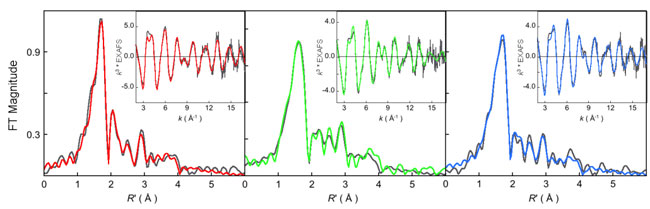
Figure 1:
k3-weighted Ni K-edge EXAFS data (inset) and their corresponding Fourier
transforms (FT). In all cases data: (gray), fit: (A)
MCRred1-silent (red),
MCRred1 (green), and MCRMe (blue).
[ large
view of figure]
The active site of MCR consists of a Ni bound tetrapyrollic cofactor called
F430, which is proposed to cycle between several different oxidation
states and electronic structures during catalysis. There are two proposed
mechanism for the catalytic generation of methane by MCR, which can be
distinguished by the first step of catalysis. In mechanism I, which is based on
the crystal structure and mechanistic work with F430 model
complexes, a methyl-Ni complex is formed as an intermediate species via
heterolytic cleavage of the S-H bond of Methyl-SCoM bond. Mechanism II, which
is based on density function theory computations avoids the formation of an
energetically unfavorable methyl-Ni(III) species and proposes a pathway
involving homolytic cleavage and generation of a methyl radical and a
Ni(II)-thiolate species.
In an article published in the April edition of Biochemistry, Sarangi
and co-workers have solved the local structure of two key intermediates
involved in mechanism I, namely the catalytically active Ni(I)
MCRRed1 state and the Methyl bound Ni(III) state
(MCRMe) (which was formed by oxidative addition of MeI to the
reduced MCRRed1 state). MCR is extremely sensitive to aerobic
oxidation, which has proved very difficult to obtain crystals for x-ray
diffraction. Solution EXAFS data were available, but to low resolution. Sarangi
et al. were able to obtain high resolution EXAFS data on both the
MCRRed1 and
MCRMe states. They also obtained EXAFS data on
MCRRed1-silent, an inactive
Ni(II) form for comparison purposes. The experiments were performed on the SMB
XAS Beam Line 9-3 at SSRL.
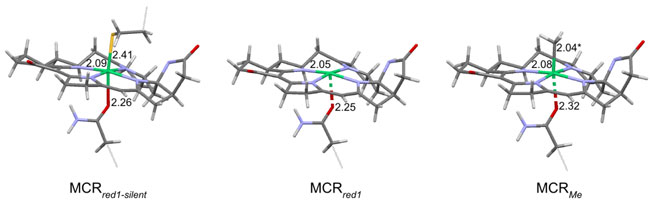
Figure 2: Schematic
diagram of the predicted active site structures of
MCRred1-silent, MCRred1, and
MCRMe based on Ni K-edge EXAFS, Ni K-pre-edge analysis and
DFT calculations.
The authors determined the local structure of the
MCRRed1 and MCRRed1-silent states
unambiguously, however, although a short axial ligand was observed in the EXAFS
data for the MCRMe state, the data were inconclusive in
determining the nature of the axial ligand. To solve this, the authors used a
combination of Ni K-edge XANES data analysis, DFT and TD-DFT calculations. The
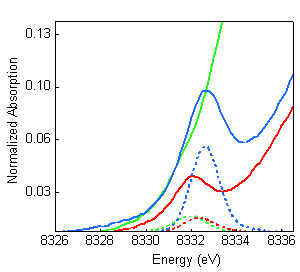
data show that the Ni K-pre-edge feature at 8331.5 eV shifts more than 1 eV to
higher energy and the intensity of the pre-edge increases almost three-fold.
These trends in pre-edge energy and intensity were reproduced by TD-DFT
calculations by only using a Ni(III)-Me model for MCRMe
(see Figure 3). Other
models (with different axial ligands) led to theoretical spectra that did not
agree with the experimental data. Together, the EXAFS, Ni K-edge XANES and DFT
results unambiguously demonstrate the presence of a unique Ni(III)-Me
electronic structure in MCRMe.
Figure 3: Comparison of
the Ni K-pre-edge XAS data (solid) with the TD-DFT calculated spectra (dashed):
MCRred1-silent (red), MCRred1 (green),
MCRMe (blue). The high intensity and energy of
MCRMe data could only be reproduced with a model with
Ni(III)-Me electronic structure. The results presented in this study indicate that the Ni-C bond in
MCRMe is long, reminiscent of the long Co(III)-C bond in
adenosylcobalamin (AdoCbl) (~2.04 Aring;). In AdoCbl-dependent enzymes, the
long weak Co-C bond undergoes a homolytic cleavage forming an Ado• radical,
which subsequently initiates radical-based substrate rearrangements while in
MeCbl, a heterolytic cleavage occurs and results in a methyl cation and Co(I).
Thus, the long Ni-Me bond in MCR observed by XAS could promote homolysis of the
Ni(III)-methyl bond, which would lead to formation of a methyl radical or
enhance the electrophilicity of the methyl group, hence increasing its
susceptibility toward nucleophilic attack. In the case of the radical
mechanism, the resulting CH3• radical can then abstract an H•
from
HSCoB, yielding CH4 and •SCoB, which subsequently would lead to the
formation of CoBS-SCoM and the active MCRred1 state of MCR.
The article also highlights an important property of the
F430 cofactor in stabilizing the uncommon Ni(I) and Ni(III)
oxidation states in biology. In the case of MCRMe the
cofactor participates in very strong covalent interaction with the Ni(III) site
thereby neutralizing the positive charge. This is consistent with the unusual
stability observed in reported biochemical studies. In the case of
MCRRed1 the low charge on a formally Ni(I) species would be
expected to increase the pKa of the coordinating anionic nitrogens and
destabilize the Ni-N bond toward dissociation and protonation. In this case the
low-lying F430 orbitals participate in back bonding interaction to
increase the charge on the Ni(I) and increase the stability of the
Ni(I)-F430 cofactor.
Primary Citation
Sarangi, R.; Dey, M.; Ragsdale, S. W. Geometric and electronic structures of
the Ni(I) and methyl-Ni(III) intermediates of methyl-coenzyme M reductase.
Biochemistry 2009, 48, 3146-3156.
References
1) Thauer, R. K. Biochemistry of methanogenesis: A tribute to Marjory
Stephenson. Microbiology, 1999 144, 2377-2406.
2) DiMarco, A. A., Bobik, T. A., and Wolfe, R. S. Unusual coenzymes of
methanogenesis. Annu. Rev. Biochem. 1990 59, 355-394.
3) Ellermann, J., Hedderich, R., Böcher, R., and Thauer, R. K. The final
step in methane formation: Investigations with highly purified methyl-coenzyme
M reductase (component C) from Methanobacterium thermoautotrophicum (strain
Marburg). Eur. J. Biochem. 1988 172, 669-678.
4) Ellefson, W. L., Wolfe, R. S., and Whitman, W. B. Nickel-containing
factor F430: Chromophore of the methyl reductase of Methanobacterium
thermoautotrophicum. Proc. Natl. Acad. Sci. U.S.A. 1982 79,
3707-3710.
{kind=link}
SSRL is supported by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.