 Rick Russell, Ian S. Millett, Sebastian Doniach and Daniel Herschlag
Rick Russell, Ian S. Millett, Sebastian Doniach and Daniel Herschlag
Stanford University
One goal of genome projects is to systematically identify genes
(1,2). The best
current knowledge indicates that humans carry approximately 35000 genes. This
number is an estimate that varies from expert to expert and range
up to 100,000 (3-5). To anyone who has taken an elementary
biology class, this ambiguity must seem strange. How hard can it be to count
genes? After all, don't cells translate genes into proteins? Counting genes
should be as easy as counting proteins.
In fact, most estimates for the number of genes in the human genome exploit
this strategy with researchers developing various ways of directly or
indirectly counting proteins (6-10). The fundamental problem
then is in the definition of a gene. A gene encodes an action - and not all
the action in a cell is in the protein.
Watson and Crick proposed a central tenet of biology over 50 years ago
(11):
DNA ® RNA ® Protein. RNA thus functioned simply as a genetic intermediary
between DNA and proteins. Even as this tenet was proposed, researchers knew
that it was at best an approximation. If RNA served only as an intermediary,
there should be some correspondence between the concentrations of amino acids
of proteins and the nucleotides of RNA and DNA. Experimentally, this was not
observed. Indeed, shortly after outlining how cells translated DNA to RNA to
proteins, Crick observed "The evidence presented there showed that our ideas
were in some important respects too simple" (12).
In 1983, Thomas Cech made a key discovery that led to his Nobel Prize. He
discovered that an RNA molecule catalyzed a structural change in a molecule; in
this case an RNA molecule (13). Until his discovery,
catalysis was a role reserved for proteins. With Cech's discovery, the
"action" happened in the RNA.
Knowledge of RNA's role within the cell has grown quickly. We now know that
RNA plays a pivotal role in gene silencing, gene shuffling, protein regulation
and even disease. As one example, researchers in Finland traced the cause of
an inherited dwarfism to a specific location in the human genome
(14). They
sequenced the entire region only to find that people with and without the
disease had exactly the same protein-encoding genes. They eventually traced
the cause of disease to differences in the space between genes. These
differences resulted in mutated 267 nucleotide non-coding RNA - RNA that did
not result in protein production in either healthy or afflicted people - that
triggered the onset of the disease.
Why did it take 30 years for someone to discover Crick's missing piece of the
biological puzzle? The problem is entirely experimental. Many of biologists'
most powerful tools do not work on RNA. For example, the Protein Data Bank
currently contains about 20,000 protein structures. In contrast, the Data Bank
contains the structures of only about 500 RNA molecules. Because of their
charged phosphate backbones, RNA molecules resist crystallization and hence it
is not possible to use x-ray crystallography to determine the 3-dimensional
structure of RNAs. Similarly, circular dichroism - a structural tool
prevalent in protein chemistry - yields little information when applied to RNA.
Until recently, plight of the RNA experimentalist was quite bleak.
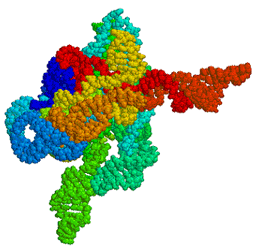
Figure
1: A 3-dimensional model of the Tetrahymena ribozyme. The primary
difference between mice and men may lie in the action of these non-coding RNAs.
In our initial studies, we used the same catalytic RNA - generically termed a
ribozyme - discovered by Cech (Figure 1).
The goal of our studies is simple: by understanding how this and other RNAs
assume a 3-dimensional shape, we hope to develop rules that describe how a
1-dimensional chain of nucleotides spontaneously takes on a biologically active
3-dimensional shape. In protein studies and genomics, this is referred to as
"solving the second genetic code", a problem of paramount importance in basic
biophysics as well as having very practical applications like drug discovery.
As our initial question, we asked what drove the folding of this ribozyme. For
example, collapse of the RNA coil might be driven by "dielectric screening".
Concentrations of salt might mask the charges on the RNA phosphates and allow
the RNA chain to compact. From biochemical work, we knew that the RNA enzyme
only catalyzed reactions in the presences of Mg2+.
Experimentally, equating shape formation with the rate of catalysis is
problematic. The ribozyme might be folded or nearly folded without
magnesium yet still not catalyze a reaction.
To test this, we looked at the structure of the ribozyme in varying
concentrations of salts. We compared our measurements to the SAXS profile of
the fully catalytic ribozyme, a structure achieved in low concentration of
MgCl2. Our results, summarized in Figure 2, show that folding of
the ribozyme
is driven by divalent cations (catalysis is specific to Mg2+).
Even if we
account for charge differences, Mg2+ is at least 25 times more
effective than Na+ in inducing structure in this ribozyme.
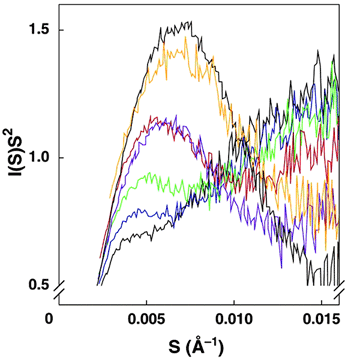
Figure 2: Gradations of
folding induced by varying salt concentrations as monitored by SAXS. Kratky
plots have the form of I(S)S2 against S, where I(S) is the
scattering intensity
and S is related to the scattering angle. In a Kratky plot, an inverted
parabola indicates a well-folded structure. More pronounced peaks indicate more
folded structures. A plot with a tail rising "to the right" (increasing
x-axis) indicates unfolding. Kratky plots are shown for the standard L-21
ScaI ribozyme at 25°C in buffer containing 20 mM Na+
(black), 70 mM Na+ (blue),
120 mM Na+ (green), 420 mM Na+ (red), 820 mM
Na+ (purple), the intermediate
Itrap (orange, formed at 15°C with Mg2+), and the
native ribozyme in buffer containing 15 mM Mg2+ (top curve -
black). [S = 2sinq/l,
where l is the x-ray
wavelength, 1.54 Ĺ, and 2q is the scattering angle].
As RNA folds to its functional form, it must undergo compaction from a
disordered chain to a specific structure. We used stopped-flow and continuous
flow kinetics in conjunction with SAXS to directly monitor the compaction
during folding of the Tetrahymena ribozyme over a time window of more
than five orders of magnitude. We wanted to determine whether compaction
occurs as an early step in folding, before specific tertiary structure
formation, or whether it occurs as long-range contacts form, which by their
nature necessitate collapse.
We observed substantial compaction in the low millisecond timescale, with
overall compaction and global shape changes largely complete within one second.
The earliest detected tertiary structure is formed at least 5-fold slower.
Thus, compaction largely precedes specific tertiary structure formation,
indicating that a nonspecifically collapsed intermediate is formed and
transiently accumulates during folding (15). We must study
other ribozymes to determine if this collapsed intermediate is a general
feature in RNA structure formation.
We note that this "molten globule" or collapsed intermediate is not universally
observed in proteins. When observed, protein molten globules are compact and
almost entirely unstructured. The individual amino acids of the protein move
about rapidly within the globule like disturbed bees in a hive. The helices
and sheets that make up the structure of proteins are absent. In proteins, the
creation of short-range secondary structure and the formation of long-range
folded structure seem to coincide. In contrast, RNA secondary structure is
formed in preparing the initial state (Figure 2) while final compaction is
triggered by addition of magnesium.
If secondary structure is formed in the initial unfolded state, what impact
does this structure have on folding? For example, one might simplistically
argue that, since high concentrations seem to induce some secondary structure,
the structure is already partially folded and would thus reach a folded
"end-state" more quickly. Conversely, if the structure induced by salt was
somehow incorrect, it might take more time for the ribozyme to assume the
correct final shape.
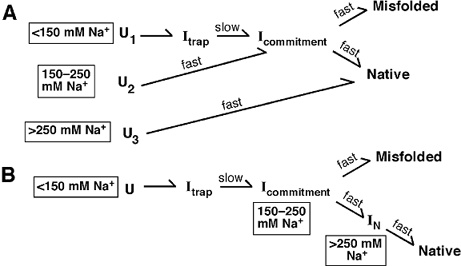
Two potential folding schemes, built from our and other authors' suggestions,
are depicted in Figure 3. Scheme B follows from the simple suggestion above:
increasing salt moves the ribozyme closer to the native folded form. Thus, at
low salt, the ribozyme starts from an unfolded state. On addition of
Mg2+, it
moves to a long-lived intermediate termed Itrap, then to another
intermediate Icommitment, and then either successfully folds or
becomes trapped in an incorrect, misfolded structure. At higher salt
concentrations, the ribozyme in Scheme B avoids the long-lived intermediate
Itrap and folds more quickly.
Scheme A is much more complicated: the path the ribozyme takes when it folds
depends on its starting state. At low salt, the pathway mimics that of Scheme
B. At intermediate concentrations of Na+, the ribozyme's starting
state is still unfolded but now has more secondary structure. On addition of
magnesium, this path skips the trap species and moves to a final, potentially
misfolded state. At still higher salt, the ribozyme folds directly to native
species.
The folding of the ribosome follows Scheme A. The argument for this is in part
based on the Kratky plots of Figure 2. We can prepare the long-lived
intermediate Itrap. This intermediate is smaller than the unfolded
ribozyme but larger than the misfolded or native final forms. We expect the
species in Scheme B to have sizes intermediate between Itrap and
the final states. Experimentally, this is not observed.
Figure
3: Two competing models for how the Tetrahymena ribozyme assumes a
final 3-dimensional state (Native). In Scheme A, different Na+
concentrations cause three different starting states to be differentially
populated. These different
starting states - U for unfolded - then fold along discrete pathways through
the landscape. With low starting concentrations of Na+, a
significant fraction (0.55) starts folding from U1. These molecules
proceed through the late folding intermediate Itrap and then
slowly escape this intermediate to partition between the native and long-lived
misfolded forms. At intermediate Na+, folding
starts from U2; Itrap is avoided but the partitioning
between the native and long-lived misfolded forms is the same as at low
Na+. This identical partitioning suggests the presence of the
common folding intermediate Icommitment. With high starting
Na+, U3 is populated and essentially all of the
ribozyme folds fast and correctly; thus, both Itrap and
Icommitment are avoided. In Scheme B, only one folding pathway is
possible. Increasing salt in the starting unfolded state moves the ribozyme
forward toward the final folded state.
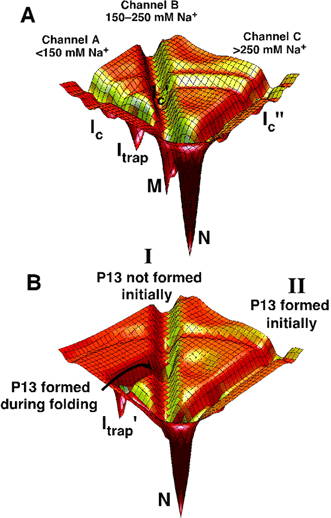
Structured RNAs achieve their active states by traversing complex,
multidimensional energetic landscapes. By probing the folding landscape of the
Tetrahymena ribozyme with single molecule fluorescence and SAXS, we have shown
that the landscape contains discrete folding pathways. These pathways are
separated by large free-energy barriers that prevent interconversion between
them, indicating that the pathways lie in deep channels in the folding
landscape (16) (Figure 4). In this, RNA structure
formation differs drastically from that seen in proteins. In proteins, the
starting state of unfolded protein has a comparatively small impact on the
folding path and final folded form.
Figure 4:
RNA folding pathways contained in channels. (A) A schematic of channels
for the ribozyme. The dependence of folding properties
Unfortunately, we have also learned that RNA folding is highly path dependent.
The observed intermediate and even the final folded product can depend on the
initial conditions or, equivalently, the structure of the starting state.
Ironically, correctly predicting the 3-dimensional structure of an RNA molecule
may hinge on choosing the right shape for the unfolded "random coil".
This research was carried out at the Stanford Synchrotron Radiation
Laboratory,
a national user facility operated by Stanford University on behalf of the U.S.
Department of Energy, Office of Basic Energy Sciences.
We have exploited small angle X-ray scattering (SAXS) to address this challenge
(15-17). In many respects, our efforts represent an ideal
marriage of a
problem and technique. SAXS provides a quantitative measure of structure
formation and conformational change, information typically not available in the
field of RNA research. RNA, in turn, has properties that make it an excellent
material for SAXS investigation. SAXS works by scattering off the difference
in electron density between solvent (water) and the biological molecule. This
"contrast" is relatively low for proteins, molecules composed primarily of
C/N/O. RNA, with its electron-rich phosphate backbone, yields much better
contrast (and data) at comparatively low concentrations. With polymerase chain
reactions (PCR), designing systems to test an idea or take measurements is
relatively straightforward. Finally, radiation induced aggregation - which can
be a significant problem in SAXS experiments, appears largely lacking with RNA.
on initial conditions indicates
the presence of free energy barriers between the channels, whereas the figure
is contoured by internal free energy. At least some of these barriers are
likely to be present when considering internal free energy because
interconversion between the starting states requires base pairs to exchange.
Therefore, "walls" are shown separating the channels. The simplest model is
shown, in which there are three pathways, A-C, that lie in channels A, B, and
C, respectively. Pathways A and B predominate at low initial Na+
concentration, pathway B at intermediate Na+ concentration, and
pathway C at high Na+ concentration. The fast-folding fraction
under
low Na+ conditions could instead arise from an additional
pathway that branches from pathway A during folding before the formation of
Itrap (not shown). Ic is a collapsed intermediate that
forms along pathway A and analogous intermediates are postulated along pathways
B and C. (B) Effects of P13 formation - a specific domain of the
ribozyme
- during folding starting with a preformed P3. The landscape for
folding of the U273A ribozyme is depicted for simplicity.
(See references for details)
Taken together, these two studies have significant implications to our goal of
transforming a 1-dimensional chain of RNA into a biologically relevant
3-dimensional shape. First, we have found that RNA forms a nonspecifically
collapsed intermediate(s) and then searches for its tertiary contacts within a
highly restricted subset of conformational space. This observation is a boon
computationally. By forming "islands" or intermediates in the folding/search
process, we can significantly speed the progress of our structural algorithms.
References
SSRL Highlights Archive