

Kumar Singh Saikatendu1, Jeremiah S. Joseph1, Vanitha
Subramanian1, Benjamin W.
Neuman3, Michael J. Buchmeier3, Raymond C.
Stevens2 and Peter Kuhn1.
1Departments of Cell Biology, 2Molecular Biology, and
3Neuropharmacology, The
Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California
92037
Severe Acute Respiratory Syndrome (SARS) is a debilitating pneumonia-like
disease of the upper pulmonary tract in humans whose causative organism is a
group II coronavirus (SARS-CoV). An early event in infected cells is the
transcription of its genomic RNA into a large (~7000 residues) replicase
polyprotein, which is then cleaved into 16 mature proteins known as
non-structural proteins or nsps. Several groups have begun to characterize
these proteins that form integral components of the SARS
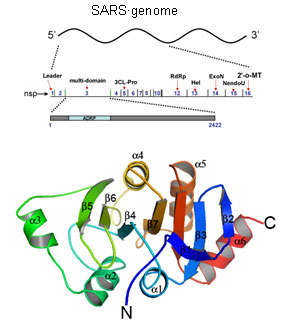
replicase/transcriptase complex. The third of these nsps, (nsp3) is a large
213kDa multidomain protein that arises due to cleavage of the polyprotein by
the virally encoded papain-like protease or PLpro. It has at least six
structural domains apart from a transmembrane domain.
We have embarked on an NIAID funded program that aims to structurally and
functionally characterize all the protein domains of the SARS proteome
(sars.scripps.edu). The program leverages recent advances made in
high-throughput structure determination pipelines like parallel 96-well format
cloning, expression and purification of proteins, nanovolume crystallization
and remote screening and data collection facilities at synchrotron beamlines.
Advances made in robotics and automation at beamlines like the 1-5, 11-1 and
11-3 at the Stanford synchrotron radiation labs (SSRL) and integrated tools
like BLU-ICE enable rapid screening of several hundred crystals remotely. A
cherished goal of this effort is to use these technologies to rapidly
characterize proteomes of emerging disease causing organisms and provide
structural templates for the structure-based design of inhibitory molecules.
One of the first structures to result from this effort was the second domain of
SARS nsp3. The structure was solved by single-wavelength anomalous dispersion
(SAD) method using data collected at the absorption edge of selenium, a
commonly employed method in high-throughput structure determination. Native
crystals diffracted to 1.4 Å at SSRL beamline 11-1 providing a very high
resolution structural model.
The structure revealed a macro-domain-like fold that is commonly seen
in a unique class of phosphatases that specifically dephosphorylate to
ADP-ribose-1"-Phosphate (Appr). The structure, its organization as a dimer in
the asymmetric unit, distribution of electrostatic charge on its surface, the
presence of a deep solvent exposed cleft and the conservation of the residues
at its putative active site immediately led to experimental validation of this
domain as a functional phosphatase specific for Appr substrate and
provided important insights into molecular aspects RNA processing and
maturation that the virus employs during its replication cycle.
Several other proteins from this virus are in different stages of the structure
determination pipeline with the ultimate goal of developing a detailed
molecular structure-function-interaction map between the different protein and
nucleic-acid components of the SARS proteome and their human cellular
counterparts.
This study was supported by NIAID/ NIH Contract # HHSN 266200400058C
"Functional and Structural Proteomics of the SARS-CoV" to Peter Kuhn.
Primary Citation
Saikatendu KS, Joseph JS, Subramanian V, Clayton T, Griffith M, Moy K,
Velasquez J, Neuman BW, Buchmeier MJ, Stevens RC, Kuhn P (2005). Structural
basis of severe acute respiratory syndrome coronavirus ADP-ribose-1''-phosphate
dephosphorylation by a conserved domain of nsP3.
Structure. 13, 1665-1675.
References
| SSRL is supported by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. |