
|  |
|
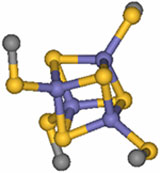
Figure 1.
Schematic repre-sentation of the common active-site iron-sulfur cluster
structural motif.
| |
Proteins containing Fe4S4 iron-sulfur clusters are ubiquitous in nature and
catalyze one-electron transfer processes. These proteins have evolved into two
classes that have large differences in their electrochemical potentials: high
potential iron-sulfur proteins (HiPIPs) and bacterial ferredoxins (Fds). The
role of the surrounding protein environment in tuning the redox potential of
these iron sulfur clusters has been a persistent puzzle in biological electron
transfer [1]. Although HiPIPs and Fds have the same iron sulfur structural motif
- a cubane-type structure - (Figure 1), there are large differences in their
electrochemical potentials. HiPIPs react oxidatively at physiological
potentials, while Fds are reduced. Recently, sulfur K-edge x-ray absorption
spectroscopy (XAS; measured at SSRL beam line 6-2) has been used to uncover the
substantial influence of hydration on this variation in reactivity in a
collaborative effort led by Stanford Chemistry and Photon Science researchers
Edward I. Solomon, Keith O. Hodgson and Britt Hedman [2].
|  |
|
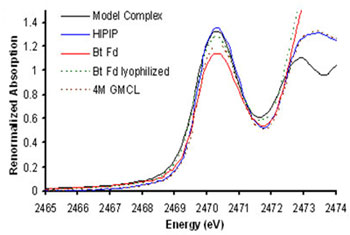
Figure 2.
Pre-edge region of the Sulfur K-edge XAS data for a model (black), wild-type
C.
vinosum HiPIP (in blue), unfolded C. vinosum HiPIP (in dashed brown), wild-type
B. thermoproteolyticus (Bt) Fd (in red) and lyophilized Bt Fd (in dashed
green).
| |
The sulfur-K XAS pre-edge involves excitation of S 1s electrons to the
unoccupied valence orbitals formed by interaction of the Fe 3d orbitals with
the S 3p orbitals [3]. Since the transition is localized on the sulfur, the
pre-edge provides a direct measure of S 3p character in the metal d orbitals
(i.e. covalency). In this study, which follows several years of developing the
methodology behind this approach [4], S K-edge XAS was used to examine the
changes in covalency in native and perturbed (lyophilized Fd and unfolded
HiPIP) protein environments. These experiments show that the Fe-S covalency is
much lower in natively hydrated Fd active sites than in HiPIPs, but increases
upon water removal (Figure 2). Similarly, HiPIP covalency decreases upon
unfolding, exposing an otherwise hydrophobically shielded active site to water
(Figure 3). These results demonstrate that Fe-S covalency is a direct
experimental marker of the local electrostatics due to H-bonding. Studies on
related model compounds and accompanying density functional theory (DFT)
calculations support a correlation between Fe-S covalency and ease of
oxidation, which suggests that differential hydration accounts for most of the
difference between Fd and HiPIP reduction potentials. This raises the
intriguing possibility that oxidation/reduction potentials can be regulated by
protein/protein and protein/DNA interactions that effect cluster hydration.
|  | |
|
Figure 3.
Schematic representation of the iron-sulfur cluster reduction potential tuning
by desolvation.
| | |
Primary Citation:
A. Dey, F. E. Jenney, Jr., M. W. W. Adams, E. Babini, Y. Takahashi, K.
Fukuyama, K. O. Hodgson, B. Hedman and E. I. Solomon, "Solvent Tuning of
Electrochemical Potentials in the Active Sites of HiPIP Versus Ferredoxin",
Science 318, 1464 (2007)
References:
-
K. Fukuyama, Handbook of Metalloproteins; A. Messerschmidt, R. Huber, T.L.
Poulos, K. Wieghardt, Eds, John Wiley & Sons, Ltd, pp. 543 (2001).
-
A. Dey, F.E. Jenney Jr., M.W.W. Adams, E. Babini, Y. Takahashi, K. Fukuyama,
K.O. Hodgson, B. Hedman, E.I. Solomon, Science 318, 1464 (2007).
-
T. Glaser, B. Hedman, K.O. Hodgson, E.I. Solomon, Acc. Chem. Res.
33, 859
(2000).
-
B. Hedman, K.O. Hodgson, E.I. Solomon, J. Am. Chem. Soc. 112,
1643 (1990); E.I. Solomon, B. Hedman, K.O. Hodgson, A. Dey, R.K. Szilagyi, Coord.
Chem.
Rev., 249, 97 (2005).
|
| PDF
Version | | Lay Summary | |
Highlights Archive
|
| SSRL is supported
by the Department of Energy, Office of Basic Energy Sciences. The SSRL
Structural Molecular Biology Program is supported by the Department of Energy,
Office of Biological and Environmental Research, and by the National Institutes
of Health, National Center for Research Resources, Biomedical Technology
Program, and the National Institute of General Medical Sciences. |
|
