![Delocalized Molecular Orbitals of the [6Fe-6S]
Cluster of the
FeFe-Hydrogenase](FeFe_title.jpg)
Hydrogenases catalyze the stoichiometrically simple (H2 ↔ 2 H+ + 2
e-), but
energetically difficult dihydrogen uptake/evolution reactions.1 The
FeFe-hydrogenases are of great interest since they can catalyze both the
forward and the reverse reactions. Under optimal conditions a single molecule
of FeFe-hydrogenase can produce approximately 6000 molecules of hydrogen within
a second. This translates into a theoretical capacity for refueling the
hydrogen tank of the Space Shuttle within 30 minutes. Thus, hydrogenases are
considered desirable biological targets for hydrogen-based energy production
and storage technologies.
| 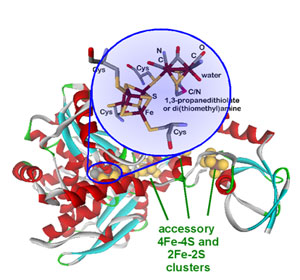 |
|
Figure 1. Molecular structure of FeFe-hydrogenase from Chlostridium
pasteurianum I 2 with H-cluster (catalytically active site) and accessory
clusters.
|
Protein crystallography revealed the molecular structure of the enzyme (Figure
1),2,3 which is composed of a
single amino acid chain with multiple iron-sulfur clusters. The catalytically
active cluster (H-cluster) shows a remarkable molecular structure with unique
organometallic character. The hydrogenases are the only group of metalloenzymes
that contain carbon monoxide and cyanide ligands which are generally toxic to
living organisms; however, they are absolutely required for hydrogenase
activity. The accessory iron-sulfur clusters are thought to be important in
efficient electron transfer to/from the H-cluster.4
Several spectroscopic techniques have already been applied to understand the
structure of this unique cluster and due to overlapping spectral features from
the accessory iron-sulfur clusters it has been difficult to determine the
physico-chemical properties of the H-cluster that are responsible for its high
catalytic activity.
Biomimetic chemistry provides an alternative way to probe the structural
properties of the H-cluster that is not be possible from direct experiments on
the protein samples. We have utilized structurally analogous iron-sulfur
clusters5 to gain insight into the electronic
structure of the H-cluster by X-ray absorption spectroscopy (XAS). In the 2-5
keV energy range (accessible at beamline 6-2 of SSRL), spectral features
observed below the ionization threshold of the sulfur 1s core electrons are
directly related to the nature and strength of chemical bonds between sulfur
atoms and transition metal ions.6 The energy
positions and intensities of these features are characteristic of the type and
strength of specific metal-sulfur interactions, respectively. The assignment of
spectral features is commonly aided by using structurally well-defined model
complexes. Spectroscopically calibrated density functional theory (DFT)
calculations7 aid in bridging the gap between the
model complexes and the ill-defined protein bound active sites.
| 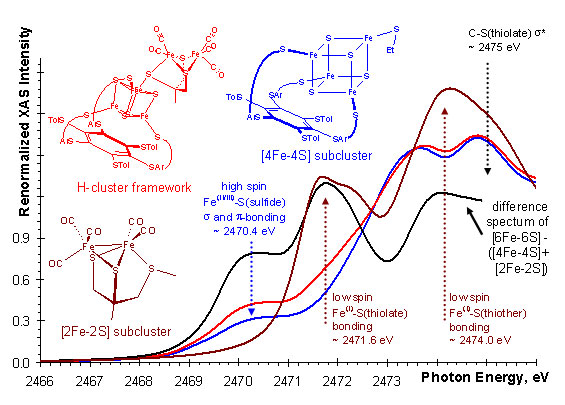 |
|
Figure 2. Comparison of sulfur K-edge spectra of 2Fe, 4Fe, and 6Fe
biomimetic models of H-cluster and the difference spectrum.
|
Using XAS in combination with DFT, the strength of the chemical interaction was
probed between the [4Fe-4S] and the [2Fe-2S] subclusters of the H-cluster. By
comparing the spectra of each subcluster and the H-cluster framework, we found
evidence (Figure 2) for considerable electron delocalization between the
subclusters upon formation of the iron-bridging thiolate-iron bond. This
suggest that the H-cluster is an electronically inseparable [6Fe-6S] cluster.
Thus, the redox chemistry and substrate activation is determined by both
subclusters together and not just the [2Fe-2S] subcluster which has been the
focus of much past research. DFT calculations on separate and the combined
subclusters also show this delocalization by redox active molecular orbitals
that span the entire 6Fe-framework (Figure 3).
| 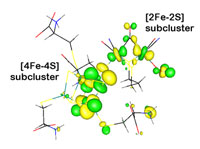 |
|
Figure 3. Redox active molecular orbital of the H-cluster showing
electron delocalization between the 4Fe- (left) and 2Fe- (right) subclusters.
|
Recently a heterologous expression system for FeFe-hydrogenases was developed,
which opened up the possibility for direct spectroscopic studies on the
metalloenzyme. An active version of the FeFe-hydrogenase can be expressed in
the absence of the accessory iron-sulfur clusters, thus reducing the background
of iron and sulfur in the XAS experiment. Further spectroscopic and
computational studies on biomimetic complexes, metalloprotein samples, and in
silico chemical models have the potential to assist in the design of
inexpensive iron/sulfur/carbonyl-based catalytic systems for hydrogen
production and conversion to electronic energy which could replace the
expensive noble metal-based systems.
Primary Citation
Schwab, D. E.; Tard, C.; Brecht, E.; Peters, J. W.; Pickett, C. J.; Szilagyi,
R. K. Chem. Commun. 2006, 3696-3698.
References
-
Adams, M. W. W. Biochim. Biophys. Acta 1990, 1020, 115.
-
Peters, J. W.; Lanzilotta, W. N.; Lemon, B. J.; Seefeldt, L. C.
Science 1998,
282, 1853-1858.
-
Nicolet, Y.; Piras, C.; Legrand, P.; Hatchikian, C. E.; Fontecilla-Camps, J. C.
Struct. Fold Des. 1999, 7, 13-23.
-
Nicolet, Y.; Lemon, B. J.; Fontecilla-Camps, J. C.; Peters, J. W. Trends
Biochem. Sci. 2000, 25, 138-143.
-
Tard, C.; Liu, X. M.; Ibrahim, S. K.; Bruschi, M.; De Gioia, L.; Davies, S. C.;
Yang, X.; Wang, L. S.; Sawers, G.; Pickett, C. J. Nature 2005, 433, 610-613.
-
Solomon, E. I.; Hedman, B.; Hodgson, K. O.; Dey, A.; Szilagyi, R. K. Coord.
Chem. Rev. 2005, 249, 97-129.
-
Szilagyi, R. K.; Winslow, M. A. J. Comput. Chem. 2006, 27, 1385-1397.
-
Posewitz, M. C.; King, P. W.; Smolinski, S. L.; Smith, R. D.; Ginley, A. R.;
Ghirardi, M. L.; Seibert, M. Biochem. Soc. Trans. 2005, 33, 102-104.
|
| PDF
Version | | Lay Summary | |
Highlights Archive
|
| SSRL is supported
by the Department of Energy, Office of Basic Energy Sciences. The SSRL
Structural Molecular Biology Program is supported by the Department of Energy,
Office of Biological and Environmental Research, and by the National Institutes
of Health, National Center for Research Resources, Biomedical Technology
Program, and the National Institute of General Medical Sciences. |
|
