 N. M. Markovi
N. M. Markovi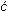 , LBNL and C. A. Lucas, University of Liverpool
, LBNL and C. A. Lucas, University of Liverpool
Ecological and political realities have moved discussions of, and advances in, fuel cell technology into mainstream public awareness. Electrocatalysis, the science of modifying the overall rates of electrochemical reactions so that selectivity, yield and efficiency are maximized, is the work from which those advances spring. Studies in electrocatalysis have resulted in highly selective multicomponent gas mixture sensors, human blood component sensors, new electrocatalysts for oxidation/reduction of inorganic and organic pollutants in air and water, as well as better electrocatalysts for the fuel cell conversion of renewable and fossil fuels to electrical work. Studies of the mechanisms by which these catalysts operate have been advanced through development of in-situ surface x-ray scattering (SXS) techniques. SXS capabilities at SSRL were recently used to investigate the interface structure of an ultrathin COad (adsorbed carbon monoxide) overlayer on platinum. This work has elevated the macroscopic description of the COad state at the solid-liquid interface to a microscopic level and enabled the relation between the reactivity and the interfacial structure of COad/Pt to be understood.
Of the various systems studied, CO oxidation [COad + OHad = CO2 + H+ + e-] on Pt and Pt-bimetallic surfaces occupies a special position in surface electrochemistry. Carbon monoxide is the simplest C1 molecule that can be electrochemically oxidized in a low temperature fuel cell at a reasonable (although not necessarily practical) potential. It thus serves as an important model "fuel" for fundamental studies of C1 electrocatalysis. For over a decade now the ability to characterize atomic/molecular spatial structures and to monitor changes in the local symmetry of surface atoms in-situ under reaction conditions has played an important part in our understanding of surface electrochemistry at metal-based interfaces [1,2]. This progress has been influenced greatly by the technique of in-situ surface x-ray scattering (SXS) which, in combination with infrared spectroscopy (IR) and electrochemical methods, has been used to find interrelationships between the microscopic surface structures of fcc metals (Pt, Ir, Pd, Au, Ag, Cu) and the macroscopic kinetic rates of the reactions. Two forms of COad species can be distinguished thermochemically on Pt(111) in an electrochemical environment [3]: (i) COad with a low heat of adsorption is characterized as the "weakly adsorbed" state, and (ii) COad with a relatively high enthalpy of adsorption is characterized as the "strongly adsorbed" state. In our recent studies, the macroscopic description of the COad state at the solid-liquid interface has been elevated to a microscopic level, which enables the relation between the reactivity and the interfacial structure of COad on Pt(111) to be understood.
Representative SXS results along with CO oxidation current in 0.05 M
H2SO4 are summarized in Figure 1. Direct information
regarding the induced relaxation of surface Pt atoms by the adsorption of CO
are obtained by analyzing and modeling crystal truncation rod (CTR) data
[4] (not shown). The potential dependence of
the Pt(111) surface relaxation, denoted as x-ray voltammetry (XRV) induced by
COad adsorption is represented by the results in Figure 1b. In
COad-free solution at 0.05 V the top Pt atomic layer expands
ca.  2 % (0.05 Ĺ) of the
lattice spacing away from the second atomic layer when the adsorbed hydrogen
(Had) reaches its maximum coverage (0.66 ML).
2 % (0.05 Ĺ) of the
lattice spacing away from the second atomic layer when the adsorbed hydrogen
(Had) reaches its maximum coverage (0.66 ML).
 |
||
|
Figure 1. (a) CO
stripping voltammetry on the Pt(111) surface in argon purged solution.
(b) ( | ||
 ) Scattering intensity changes at
(1, 0, 3.6) for the surface covered by COad;
(c) Measured x-ray intensity at (1/2,1/2, 0.2) as a function of
electrode potential for the same conditions as in (b). (d) A rocking
scan through the (1/2,1/2, 0.2) position and ideal model for the
) Scattering intensity changes at
(1, 0, 3.6) for the surface covered by COad;
(c) Measured x-ray intensity at (1/2,1/2, 0.2) as a function of
electrode potential for the same conditions as in (b). (d) A rocking
scan through the (1/2,1/2, 0.2) position and ideal model for the
Following the adsorption of CO at 0.05 V, the Pt surface expansion is even larger ca. 4 %. The difference in relaxation of the Pt(111) surface covered with Had and COad probably arises from the difference in the adsorbate-metal bonding, the Pt(111)-COad interaction being much stronger than the Pt(111)-Had interaction. At 0.05 V, no change in the relaxation of the Pt surface atoms was observed after replacement of CO from solution with nitrogen, indicating that COad is indeed irreversibly adsorbed on the Pt(111) surface. Upon sweeping the potential positively from 0.05 V, the oxidation of COad in the so-called pre-oxidation potential region (0.3 < E < 0.6 V) is mirrored with a small contraction of the Pt surface layer. Above ca. 0.6 V, the top layer expansion is reduced significantly, contracting above 0.7 V to the unrelaxed state that the Pt(111) surface has in the absence of COad.
Direct information regarding the COad structure on Pt(111) was
obtained by searching in the surface plane of reciprocal space for diffraction
peaks characteristic of an ordered adlayer. At 0.05 V, a diffraction pattern
consistent with a p(2 x 2) symmetry was observed between
0.05 < E < 0.6 V. A
rocking scan through the (˝, ˝, 0.2) position together with the derived
structural model, which consists of three CO molecules per p(2 x 2) unit cell,
is shown schematically in Figure 1d. From the width of this peak and from the
result of similar fits to other p(2 x 2) reflections a coherent domain size in
the range of 80-120 Ĺ for the CO adlayer was deduced. Upon the reversal of the
electrode potential at ca. 0.6 V, the p(2 x 2)-3CO structure is not
re-formed, Figure 1c, confirming that the structure is coverage-dependent and
not just potential-dependent.
 |
|
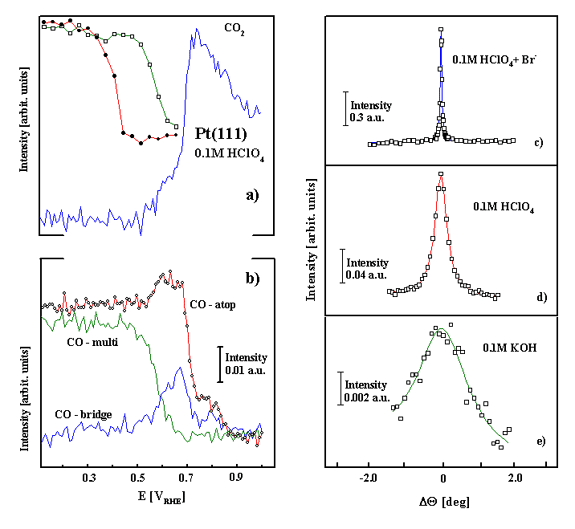
Figure 2. (a) Potential-dependent stability of the p(2x2)-3COad structure on Pt(111) in 0.1 M HClO4 in CO-saturated solution, and CO2 production as a function of electrode potential (data extracted from FTIR measurement) during the oxidation of COad. (b) Integrated intensities for COad-atop, COad-multi and COad-bridge on Pt(111) as a function of electrode potential in CO-saturated 0.1 M HClO4 solution. A rocking scan through the (1/2,1/2,0.2) position for the p(2x2)-3COad structure formed at 0.05 V Pt(111) in: (c) 0.1 M HClO4 + 10-3 M Br-; (d) 0.1 M HClO4; and (e) 0.1 M KOH. |
With a constant overpressure of CO in the x-ray cell, the SXS experiments
revealed a reversible loss and re-formation of the p(2 x 2)-3CO
structure, with the p(2 x 2)-3CO structure re-forming as the potential was
slowly (1 mV/s) swept below 0.2 V, Figure 2a. IR has provided a valuable
complement to the structural information obtained from SXS [3]. As shown in Figure 2b, the
spectra for COad on Pt(111) in CO-saturated 0.1 M HClO4
have three characteristic Pt-CO stretching frequencies, the atop CO near 2070
cm-1, the multi-coordinated COad near 1780
cm-1, and the bridge COad near 1840 cm-1.
Figure 2 shows that oxidation of the COad in the three-fold hollow
sites and relaxation of the remaining COad into bridge sites and
atop sites is accompanied with both the decrease in the Bragg peak intensity
for the p(2 x 2)-3CO structure and CO2 formation. Therefore, the
macroscopic characterization of a weakly adsorbed state is linked
microscopically to a saturated CO adlayer consisting of three COad
molecules per p(2 x 2) unit cell located in a-top and three-fold hollow sites
of the Pt(111) surface. Figure 2
shows that oxidation of the COad in the three-fold hollow sites and
relaxation of the remaining COad into bridge sites and atop sites
disrupts the long-range ordering in the remaining adlayer, as the Bragg peak
intensity for the p(2 x 2)-3CO structure decreases rapidly in this potential
region. The "relaxed" COad adlayer (characterized microscopically)
with substantial alternation in binding site geometry from predominantly
three-fold hollow to bridge sites can be linked macroscopically to the
strongly adsorbed state of COad that is
oxidized in the ignition potential region. It is also worth mentioning that
besides tuning the fine balance between atop and multifold coordinated
COad, the domain size of the COad structure is
significantly affected by the nature of anions. For example, the domain size
(Figures 2d-2f ) and stability (not shown) of the p(2 x 2)-3CO structure
increases from KOH (ca. 30 Ĺ), to HClO4 (ca. 140 Ĺ)
to HClO4 + Br- (ca. 350 Ĺ); i.e., the
less active the surface is towards COad oxidation, the larger the
ordered domains of the
p(2 x 2)-3 CO structure. As discussed in reference
[3], a self-consistent explanation for this result is that
both the stability and domain size of the ordered COad adlayer are
determined by the competition between OHad and spectator anions for
the defect/step sites on the Pt(111) surface.
Acknowledgment
This work was supported by the Assistant Secretary for Conservation and
Renewable Energy, Office of Transportation Technologies, Electric and Hybrid
Propulsion Division of the U.S. Department of Energy under Contract No.
DE-AC03-76SF00098. Research was carried out in part at SSRL which is funded by
the Division of Chemical Sciences(DSC), U.S. DOE. CAL acknowledges the support
of an EPSRC Advanced Research Fellowship.
References
-
C. A. Lucas and N. M. Markovi
 , in 'Encyclopedia of Electrochemistry',
Volume 2
, in 'Encyclopedia of Electrochemistry',
Volume 2
(Wiley-VCH, 2003) Chapter 4. - N. M. Markovi and P.N. Ross, Surf. Sci. Rep., 45 (2002)
121-229.
- N.M.
Markovi, C.A. Lucas, A. Rodes, V. Stamenkovi, P.N.
Ross, Surf. Sci., 499 (2002), L149-L158.
- C. A. Lucas, N.M. Markovi, P.N. Ross, Surf. Sci., 425
(1999) L381-L386.
SSRL Highlights Archive