

The glial cell line-derived neurotrophic factor (GDNF), neurturin (NRTN),
artemin (ARTN), and persephin (PSPN) are GDNF family ligands (GFLs) that are
crucial for the development and maintenance of many neurons [1,2]. The trophic effect of GFLs on the
dopamine and motor neurons has stimulated interest in their use for the
treatment of neurodegenerative diseases such as Parkinson's. These structurally
related neurotrophic factors signal by forming a ternary complex with a
nonsignaling, ligand-specific GFRa receptor and a
signaling and shared receptor tyrosine kinase RET. Four different GFRa receptors (GFRa1-4) have
been identified. The preferential interactions between GFLs and GFRa receptors have also been established as GDNF to
GFRa1, NRTN to GFRa2,
ARTN to GFRa3, and PSPN to GFRa4 [3]. Given the importance of
GFLs in basic neurobiology and their potential therapeutic value, it is a
compelling goal to understand the molecular basis of the interactions between
GFLs and their receptors.
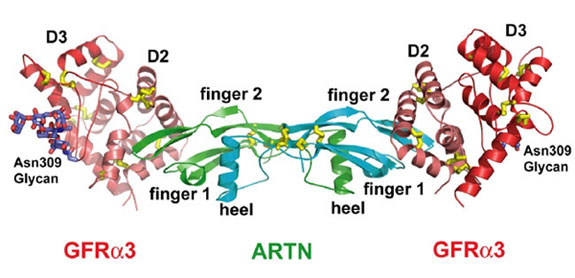
Figure 1. Overall structure the ARTN-GFRa3
complex in ribbon representation. One ARTN homodimer (monomers in cyan and
green) binds two truncated GFRa3 receptors (D2 in deep salmon and D3 in red).
The observed N-linked carbohydrates at Asn-309 position of GFRa3 are shown as
sticks in dark blue. (From Wang et al., 2006)
The structures of ARTN-GFRa3 binary complex and
unbound ARTN in two crystal forms have been determined by a combination of
heavy atom and molecular replacement methods using data collected at SSRL Beam
Line 11-1 and at the ALS. The binary complex is composed of one ARTN homodimer
and two truncated GFRa3 receptors consisting of the
D2 and D3 domains (Figure 1). Instead of being two independent domains as
people previously thought, the D2 and D3 domains were packed together to form a
globular structure. Both D2 and D3 domains are folded as a triangle spiral,
having disulfide bonds in the corners of the triangle to fix the fold. The ARTN
monomer structure has two b sheet fingers, a
cysteine-knot core motif, and an a-helical heel. Two
ARTN monomers form a symmetric homodimer with an inter-chain disulfide bond.
Figure 2. Ligand-receptor contacts between Artemin and
GFRa3. (A)
Molecular surfaces highlight the knob-in-hole complementarity between the
protruding ARTN finger region (green and cyan) and the recessed center of
GFRa3
D2 domain (deep salmon) formed by helices a1,
a2, and a5.
(B) Residues in the
common anchor points are colored in blue, and potential binding specificity
determinants are colored in green on a background of the total buried surface
(light brown). The same colors are applied to the residues in the sequence
alignment. (From Wang et al., 2006)
The complex structure of ARTN with GFRa3 revealed a
convergent recognition mode for all GFLs. In the ARTN/GFRa3 binding interface, the tip ends of fingers 1 and 2 of
ARTN insert into a pocket in the center of GFR 3 D2 domain surrounded by
helices a1, a2, and
a5 (Figure 2a). The ARTN/GFRa3 interface has two contact patches in the center, one
hydrophobic and one hydrophilic, which are conserved in all GFL-GFRa pairs. The hydrophobic patch is composed of residues
Met-199 and Trp-205 of ARTN and Tyr-182, Gly-183, and Ala-236 of
GFRa3. All
these positions are conserved as hydrophobic residues in other GFLs and
GFRa
receptors (Figure 2b). Residues Glu-143 of ARTN and Arg-179, Arg-230 of
GFRa3
in the hydrophilic patch are strictly conserved in all
GFL-GFRa pairs (Figure
2b). Mutations of the conserved positions in GDNF resulted in a completed loss
of its binding activity for GFRa1 receptor
[4]. While these residues clearly
serve as common anchor points, the surrounding non-conserved residues may be
responsible for the binding specificity between GFLs and
GFRa receptors.
Based on the complex structure and other information, we have proposed two
composite RET binding surfaces on the ARTN-GFRa3
binary complex, which would facilitate the recruitment of two RET receptors,
leading to the close proximity of RET intracellular tyrosine kinase domains
required for the signaling.
This work was supported by NIH grant RO1 HL077325, The Keck Foundation, The
Christopher Reeve Paralysis Foundation, and The Howard Hughes Medical
Institute.
Primary Citation
References

Wang, X., Baloh, R.H., Milbrandt, J., and Garcia, K.C. (2006). Structure of
artemin complexed with its receptor GFRa3:
Convergent recognition of glial cell line-derived neurotrophic factors.
Structure 14, 1083-1092.
| SSRL is supported by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. |