 by Thiang Yian Wong, Robert Schwarzenbacher and Robert C. Liddington
by Thiang Yian Wong, Robert Schwarzenbacher and Robert C. Liddington
The Burnham Institute, 10901 N Torrey Pines Rd, La Jolla, CA 92037, U.S.A.
Anthrax Toxin, together with its bacterial capsule, is a major virulence factor in Anthrax1. The virulent strain of Bacillus anthracis is an encapsulated gram-positive, rod-shaped, spore-forming bacterium that produces and exports the three Anthrax Toxin proteins, Protective Antigen (PA), Lethal Factor (LF) and Edema Factor (EF). The infectious agent of anthrax, the bacillus spore, may be introduced into a mammalian host through inhalation, ingestion and subcutaneous wounds. Once in a susceptible host organism, the spores germinate within macrophages and their vegetative cells proliferate rapidly2. The
 | |
|
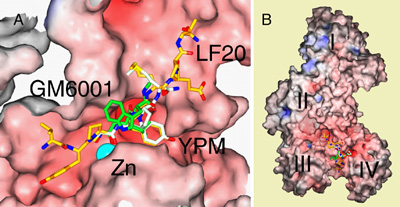
Figure 1. Crystal structures of LF-LF20, LF-thioacetylYPM-zinc, and
LF-GM6001-zinc complexes.
The molecular surface of LF is colored by charge (red = negative; blue = positive), with Zn2+ shown as a solid sphere (cyan) and the model of the peptide or inhibitor shown in stick representation. (A) The superposed individual complex structures of the three target molecules: LF20 (yellow gold) in the absence of Zn2+, resolution limit 2.85 Ĺ; SHAc-YPM (tinted white, labeled "YPM"), resolution limit 3.50 Ĺ; and GM6001 (green), resolution limit 2.70 Ĺ. The model of bound LF20 shows the sequence VYPYPMEPT (residues 8 to 16 of the 20-residue long LF20). This was the ordered region, and the electron density was clearly visible in difference maps (2Fo-Fc and Fo-Fc) calculated from crystal X-ray diffraction data. All three molecules are shown bound in the substrate-binding groove of LF, using the surface calculated for LF-LF20. The targets are all bound in the same N to C peptide orientation. (B) An overview of LF bound to the targets LF20, GM6001, and SHAc-YPM, superposed and colored as in (A). The molecular surface was calculated from the LF-LF20 complex. The domains in LF are labeled I, II, III and IV. The catalytic site is in Domain IV, where the zinc atom (not seen in this figure) is bound. These figures were prepared using SPOCK14. | |
 | |
|
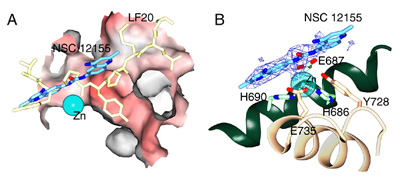
Figure 2. X-ray crystal structure of the LF-NSC 12155-Zn complex.
(A) Molecular surface of LF colored by charge (red = negative; blue = positive), with Zn2+ as a solid sphere in cyan. The model of the inhibitor molecule NSC 12155 (carbons are in light blue), and an optimized peptide with the sequence VYPYPME in light yellow, both shown in stick representation. The inhibitor NSC 12155 binds to LF in the active site at the hydrophobic neck of the pocket, where Tyrosine 4 of sequence VYPYPME (equivalent of Tyrosine 11 of LF20) is highly selected as the optimized residue to bind in this position. The binding position of NSC 12155 overlaps that of the optimized peptide sequence at residues 2 and 3 (YP). This figure was prepared using SPOCK14. (B) The inhibitor NSC 12155 bound in the active site of LF. The difference map, 2Fo-Fc, calculated at 2.9-Ĺ resolution limit, is contoured at 1.0 s. A portion of NSC 12155 appears non-rigid owing to a rotatable bond, with patchy electron density trace at 1.0 s level, although almost full electron density coverage is seen for this portion at a contour level of 0.6 s. Inhibitor molecule (light blue), zinc-coordinating residues (H686, H690, E735) and catalytic residues (E687, Y728) are in stick representation. The Ca atoms of residues 680-694 (green, background) and 726-742 (beige, foreground) are in ribbon representation. The Zn2+ ion (cyan) is a lined sphere, and its hydrogen bonds with His686, His690 and Glu735 are represented as aligned small white spheres. These figures were prepared using SPOCK14 (http://mackerel.tamu.edu/spock/). | |
In the work with colleagues at Harvard Medical School, the structural basis for LF target recognition was investigated initially through finding an optimized substrate for LF10. Two sets of peptide libraries with randomized amino acid sequence in 13 residue-long peptides were generated, with either the sequence N-terminal or C-terminal to the peptide cleavage site randomized. Each residue that contributed to increased substrate catalysis rate (increased turnover) at the individual specific position on the peptide substrate was scored. A consensus peptide sequence was built upon these results and we designed an optimized peptide substrate for further experimental work. A 20-residue long peptide was synthesized, using the consensus 13-residue sequence identified, flanked by the amino acid sequence found in the natural substrate for LF, MAPKK2. This peptide was named LF20 (MLARRKKVYPYPMEPTIAEG). LF20 provided a probe to illustrate the shape, extent and limits of the substrate-binding and catalytic site of LF. Based on the consensus sequence, a small peptidic inhibitor named SHAc-YPM, consisting of a zinc-binding thioacetyl moiety coupled to the three residues C-terminal to the cleavage site in LF20, was also created (Ki = 11 ± 3 µM). Looking at other known metalloprotease inhibitors with similar peptidic sequences to SHAc-YPM, GM600112, 13, a commercially available potent inhibitor of matrix metalloproteases (MMPs, a family of cell surface proteins in mammalian cells implicated in cancer tumour growth), MMP1 and MMP2, was assayed for and showed significant inhibition towards LF (Ki = 2.1 ± 0.2 µM).
Simultaneously, in collaboration with colleagues at USAMRIID, who performed a
search for an effective LF inhibitor in a high-throughput format, using the
National Cancer Institute (NCI) Diversity Set
(http://dtp.nci.nih.gov/branches/dscb/diversity_explanation.html),
several candidate small molecule compounds were identified11 and set up for
LF-inhibitor complex crystals. One LF-inhibitor complex that gave the best
crystal data was that of LF bound to the hit, NSC 12155, a quinolone compound
that gave the highest inhibition constant in this screen
(Ki = 0.5
± 0.18 µM).
Crystal structures of LF-LF20, in the absence and presence of zinc10, and LF-SHAc-YPM10, LF-GM600110 and LF-NSC1215511, all in the presence of zinc, were obtained to explore the three-dimensional relationship between LF and its binding targets. The striking features that were evident from the crystal structures were that all of the peptides and small molecule inhibitors recognized and bound to LF via the hydrophobic substrate-binding groove in the immediate vicinity of the catalytic zinc. LF20 induced a conformational change in the active site of LF, and Tyr11, when bound in the S1' hydrophobic pocket in LF causes the pocket to expand, in a phenomena termed "induced fit". This would not have been known if we did not have the consensus peptide LF20 to probe the binding site of LF, and thus presents a novel look at the elasticity of LF in its active site. The small molecule inhibitors on the other hand, provided us with an indication of the criteria for inhibitor design, which guides us to conclude that the main determinants for most effective inhibition is through zinc-deprivation by a strong metal-chelating moiety, like the hydroxamate group, coupled to a hydrophobic and stereochemically well-fitted small molecule structure. The structure of LF-NSC12155 with11 and without zinc (unpublished data), indicates that it may even be sufficient to have a compound with hydrophobic interactions alone (without metal chelation) to effectively inhibit the catalytic activity of LF.
Due to the diffraction limits and large unit cell dimensions of the protein crystals possibly grown for LF, data collection at a synchrotron facility like SSRL greatly enhances our ability to obtain data of high quality and resolution to achieve the results for our studies. Crystal structures will be highly important to guide future drug-designing efforts for LF inhibitors. The continuing work on the inhibitors for LF will be towards generating chemical derivatives with better inhibitory capabilities and cell permeability for effective delivery of the potential drug molecule to the centre of LF activity in the event of anthrax intoxication. We are at present working on this aspect of our research, and will be frequent users of the SSRL for its synchrotron radiation source.
References
- Leppla SH in Comprehensive Sourcebook of Bacterial Protein Toxins 2nd edn (eds Alouf JA & Freer J) 243-263 (Academic, London, 1999).
- Hanna PC, Acosta D & Collier RJ. On the role of macrophages in anthrax. Proc. Natl. Acad. Sci. USA 90, 10198-10201 (1993).
- Bradley KA, Mogridge J, Mourez M, Collier RJ & Young JAT. Identification of the cellular receptor for anthrax toxin. Nature 414, 225-229 (2001).
- Petosa C, Collier RJ, Klimpel KR, Leppla SH & Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature 385, 833-838 (1997).
- Vitale G, Bernadi L, Napolitani G, Mock M & Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 352, 739-745 (2000).
- Duesbury NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson AK, Fukasawa K, Paull KD & Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280, 734-737 (1998).
- Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 79, 3162-3166 (1982).
- Pannifer AD, Wong TY, Schwarzenbacher R, Renatus M, Petosa C, Bienkowska J, Lacy DB, Collier RJ, Park S, Leppla SH, Hanna P & Liddington RC. Crystal structure of the anthrax lethal factor. Nature 414, 229-233 (2001).
- Drum CL, Yan S-Z, Bard J, Shen Y-Q, Lu D, Soelaiman S, Grabarek Z, Bohm A & Tang W-J. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 415, 396-402 (2002).
- Turk BE and Wong TY, Schwarzenbacher R, Jarrell ET, Leppla SH, Collier RJ, Liddington RC & Cantley LC. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat. Struct. Mol. Biol. 11, 60-66 (2004).
- Panchal RG, Hermone AR, Nguyen TL, Wong TY, Schwarzenbacher R, Schmidt J, Lane D, McGrath C, Turk BE, Burnett J, Aman MJ, Little S, Sausville EA, Zaharevitz DW, Cantley LC, Liddington RC, Gussio R & Bavari S. Identification of small molecule inhibitors of anthrax lethal factor. Nat. Struct. Mol. Biol. 11, 67-72 (2004).
- Grobelny D, Poncz L & Galardy RE. Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry 31, 7152-7154 (1992).
- Levy DE, Lapierre F, Liang W, Ye W, Lange CW, Li X, Grobelny D, Casabonne M, Tyrrell D, Holme K, Nadzan A & Galardy RE. Matrix metalloproteinase inhibitors: a structure-activity study. J. Med. Chem. 41, 199-223 (1998).
- Christopher JA. SPOCK: The Structural Properties Observation and Calculation Kit [program manual] (The Center for Macromolecular Design, Texas A&M University, College Station, TX, 1998).
SSRL Highlights Archive