 Irimpan I. Mathews1, Ashley M. Deacon1,2,
Jaume M. Canaves2,3, Daniel
McMullan2,4, Scott A. Lesley2,4, Sanjay
Agarwalla5, and Peter Kuhn1,2,6
Irimpan I. Mathews1, Ashley M. Deacon1,2,
Jaume M. Canaves2,3, Daniel
McMullan2,4, Scott A. Lesley2,4, Sanjay
Agarwalla5, and Peter Kuhn1,2,6
1Stanford Synchrotron Radiation Laboratory, Stanford, CA, 2The Joint Center for Structural Genomics, CA, 3San Diego Supercomputer Center, La Jolla, CA, 4The Genomics Institute of Novartis Research Foundation, San Diego, CA, 5University of California, San Francisco, CA, 6The Scripps Research Institute, La Jolla, CA
The huge number of complete genome sequences are fueling large-scale bioinformatics, and structural and functional proteomics efforts aimed at accelerating the identification and characterization of new drug targets, a critical pre-requisite for the development of new therapeutics. The JCSG structural genomics effort is pursuing a full proteome analysis of Thermotoga maritima, and follow-on studies have already resulted in the identification of an antibacterial drug target.
The identification of thymidylate synthase complementing protein (thyX/thy1) as a thymidylate synthase enzyme (Myllykallio et al., 2002) and its structure-based functional analysis lead to the characterization of TSCP as an antibacterial drug target (Kuhn et al., 2002; Mathews et al., 2003). In general, TSCPs complement the activity of thymidylate synthase (TS) enzymes in organisms without TS. Thymidylate synthase methylates 2'-deoxyuridine 5'-monophosphate (dUMP) to produce 2'-deoxythymidine 5'-monophosphate (dTMP), a nucleotide essential for synthesis of DNA. The cofactor 5,10-methylene-5,6,7,8-tetrahydrofolate (CH2H4folate) serves as both a methyl donor and reductant in the reaction. Thymidylate synthesis is the terminal step in the sole de novo synthetic pathway to dTMP. Consequently, TS inhibition stops DNA production, arresting the cell cycle and eventually leading to "thymineless" cell death. The medical community has long been turned to TS inhibitors as a possible source of anticancer drugs.
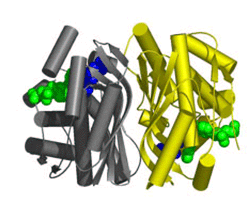
Interfering with dTMP production is a seductive strategy for drug design because it can directly prevent DNA replication. Identification of TSCP enzymes, which share no sequence or structural homology to classical TS found in humans (Figure 1), offers the possibility of therapeutic intervention with low cross-reactivity against mammalian enzymes. Furthermore, the discovery of TSCP in numerous pathogenic organisms provides an exciting opportunity for drug design.
|
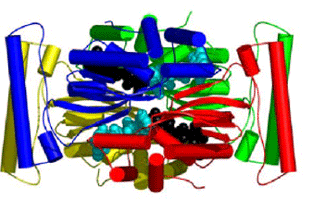 |
Figure 1. Comparison of the crystal structure of classical thymidylate synthase on the left and the structure of thymidylate synthase complementing protein. Ligand molecules bound in the active site are shown as spheres. |
|
In contrast to the classical TS enzymes, the TSCP enzymes employ CH2H4folate as the methyl donor and FAD as the reductant (Myllykallio et al., 2002). The present study with several complexes of a TSCP from Thermotoga maritima (TM0449) has located the active site and identified the residues that are important for the TSCP activity (Mathews et al., 2003). TM0449 structure shows that the enzyme is a tetramer with four identical subunits that extensively interact with each other. Two pairs of interacting active sites are located in a large groove running along the long edges of the tetramer. Multiple complexes with and without FAD, substrate and substrate analogs have mapped out many of the principal binding determinants of this novel enzyme class.
Analysis of the crystal structures in conjunction with sequence data for a number of TSCP enzymes identifies conserved residues that are important for catalysis,
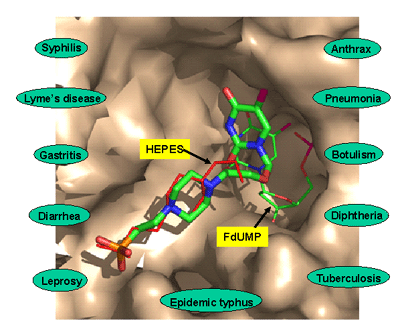 | ||
Figure 2. View of the modeled inhibitor (thick lines) and the names of disease that could be targeted by a good TSCP inhibitor. The protein atoms are shown with a surface representation. | ||
Elucidation of the function and mechanism of TSCP was enabled by the high-throughput structural genomics approach of the JCSG, which includes critical components in bioinformatics, protein production, crystallization, and synchrotron based structure determination. In his preview of the TSCP work Montfort concludes that "with TSCP, the promise of proteomics for medical advancement is beginning to be realized" (Montfort, 2003).
References
SSRL Highlights Archive