

Figure 1. The biomimetic complexes that model the OEC in the final step of water oxidation. In these complexes, a redox-active iron atom (orange) is bound to a TMC ligand (1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane, shown in gray and blue) and a peroxide moiety (red), which binds a redox- inactive metal ion (Mn+, green). Mn+ = Ca2+, Sr2+, Zn2+, Y3+ and Sc3+.
Redox-inactive metal ions play crucial roles in tuning the reactivity of oxygen-containing metal complexes and metalloenzymes such as the oxygen-evolving complex (OEC) in photosystem II (PSII) and its small-molecule mimics. In PSII, a Ca2+ ion is part of the tetrameric Mn cluster, Mn4CaO5, and is integral to the water oxidation catalysis and evolution of oxygen by the OEC. Several different functional roles have been proposed for this Ca2+ cofactor based on intense enzymatic and model studies, such as modulation of the reduction potential of the manganese center, and enhancement of the nucleophilic reactivity of water or hydroxide bound to the Ca2+ ion. Interestingly, the only other redox-inactive metal ion capable of replacing the Ca2+ center and maintaining O2 evolution is Sr2+.
To critically analyze the role of redox-inactive metal ions in metalloenzyme redox chemistry, previous studies successfully demonstrated and examined the binding of redox-inactive metal ions to high-valent metal-oxo complexes in several first-row transition metal systems. These studies demonstrated that the redox properties and, more importantly, reactivities in oxidation reactions of metal(IV)–oxo complexes are markedly affected by binding of redox-inactive metal ions. Extending studies of high-valent metal-oxo complexes to peroxide-bound non-heme Fe(III) complexes, for instance, Wonwoo Nam from Ewha Womans University (Seoul, Korea), in collaboration with SSRL researcher Riti Sarangi, demonstrated the binding of Y3+ and Sc3+ to [(TMC)FeIIIO2]+ (TMC, 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane).1 However, the binding of biologically relevant redox-inactive metals such as Ca2+ and Sr2+ was not shown.

Figure 2. EXAFS experiments and DFT geometry-optimized structures for [Fe(TMC)O2]+ bound to Ca2+ (top), Sr2+ (middle) and Zn2+ (bottom). Non-phase-shift-corrected Fourier transforms and their FEFF best-fits. Insets show the corresponding DFT geometry-optimized structures. The distances obtained from EXAFS are shown (red) for comparison with the DFT distances (black).
In the new study, published in Nature Chemistry, the researchers extended their previous analyses to include Ca2+-, Sr2+- and Zn2+- binding to [(TMC)FeIIIO2]+ (see Figure 1).2 They also investigated the chemical properties of the resulting compounds, especially their reactivity towards one-electron reduction and oxidation.
The structures of these biomimetic complexes were determined using a combination of Fe-K-edge extended x-ray absorption fine structure (EXAFS) spectroscopy at SSRL’s Beam Lines 7-3 and 9-3 and density functional theory (DFT) methods. The experimental data were consistent with a side-on-bound diamond Fe-O2-Mn+ core in all the complexes (except for Zn2+, which binds in an end-on fashion to the O2 moiety), demonstrating strong binding between Fe-O2 and the redox-inactive metal ions (see Figure 2).
The structural characterization of these molecules was complemented with reactivity studies. Based on these studies, the [FeIII(TMC)-O2-Mn+] complexes neatly divided into two groups with varying reactivities. The O2-bound intermediates were formed when O2, the redox-inactive metal ions and [(TMC)FeII]2+ reacted photochemically in the presence of a reducing agent. However, further addition of the reductant did not convert the Ca2+- and Sr2+-bound complexes into the high-valent [(TMC)FeIV(O)]2+ species. In contrast, the Y3+, Zn2+ and Sc3+ species were converted into the [(TMC)FeIV(O)]2+ via O-O bond cleavage.
Interestingly, when these intermediates - [(TMC)FeIIIO2]+ binding Ca2+ or Sr2+ - were reacted with a one-electron oxidizing agent (cerium ammonium nitrate, CAN), [Fe(TMC)]2+ was formed and the release of O2 gas (as tested by gas chromatography) was observed. In contrast, no reaction took place when the metal-ion-bound peroxide intermediates (metal ion = Zn2+, Y3+ and Sc3+) were reacted with CAN. These studies demonstrate a clear impact of the Lewis acidities of the redox-inactive metal ions on the redox potentials and reactivities of these compounds (see Figure 3).
These results can be extended to shed light on the mechanism of the oxygen-evolving center in PSII. Despite numerous studies, the mechanism of O2 evolution is still a mystery. Especially, the role of the innocent metal ion Ca2+ in the active site Mn4CaO5 cluster is not well understood. The series of compounds studied here conceptually mimic the metal-ion-bound peroxide intermediate that may form in the final steps of O2 evolution. As shown, the Lewis acidities of Ca2+ and Sr2+ tune the reactivity and redox properties of the [FeIII(TMC)-O2-Mn+] complexes to an extent where the facile release of O2 is not hindered. In contrast, stronger Lewis acids such as Zn2+ bind too tightly and hinder O2 release. Thus, the present results get one step closer to answering why nature chooses the Ca2+ ion in the oxidation of water to evolve O2 and why the Sr2+ is the only surrogate to replace Ca2+ for the reactivity of the OEC in PSII.
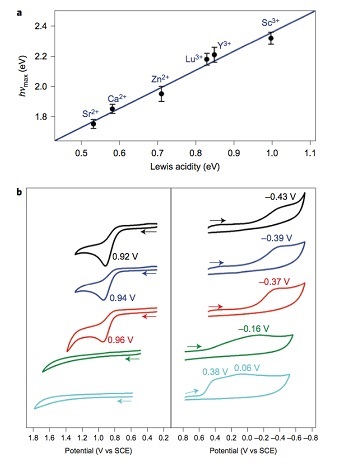
Figure 3. Effects of Lewis acidity of the redox-inactive metal ion in [FeIII(TMC)-O2-Mn+] complexes. (top) A plot of the hνmax absorption spectra versus Lewis acidity of metal ions (ΔE). (bottom) Cyclic voltammogram of the intermediates. (left) one-electron oxidation. (right) one-electron reduction.
The research was supported by KOSEF/MEST of Korea through the CRI (NRF- 2012R1A3A2048842), GRL (NRF-2010-00353), MSIP of Korea through NRF (2013R1A1A2062737) and an ALCA project from JST from MEXT of Japan. Stanford Synchrotron Radiation Lightsource (SSRL) operations are funded by the U.S. Department of Energy (DOE) Basic Energy Sciences. The SSRL Structural Molecular Biology program is supported by the National Institutes of Health National Institute of General Medical Sciences (P41GM103393) and DOE Biological Environmental Research (R.S.).
- Y.-M. Lee, S. Bang, Y. M. Kim, J. Cho, S. Hong, T. Nomura, T. Ogura, O. Troeppner, I. Ivanović-Burmazović, R. Sarangi, S. Fukuzimi, W. Nam, Chem. Sci. 4, 3917 (2013), DOI: 10.1039/c3sc51864g
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, S. Nishida, M.-S. Seo, R. Sarangi, S. Fuhkuzumi, W. Nam, Nat. Chem. 6, 934 (2014), DOI: 10.1038/nchem.2055
