
Secondary metabolites produced by microorganisms have a market value of over $30 billion annually, and nearly half of these compounds are naturally produced by bacteria in the phylum Actinobacteria1. Phylum is a taxonomic rank in biology. It is below kingdom (e.g. Animal, Plant, Fungi etc.) and above class (e.g. Mammalia). Although there are over a dozen classes of secondary metabolites, the polyketides are arguably the most versatile with medically relevant activities including antibiotic, anticancer, immunosuppressive, anti-parasitic, and cholesterol-lowering properties. As an example, approximately 4,000 tons of erythromycins (macrolide antibiotics derived from polyketides) are produced annually from the actinomycete Saccharopolyspora erythraea2. This microbe is one of many soil-dwelling bacteria that employ gigantic enzyme catalysts called polyketide synthases (PKSs) to construct complex polyketide products such as the 14-membered lactone ring of erythromycin.
The aglycone precursor of erythromycin, 6-deoxyerythronolide B, is synthesized by the prototypical polyketide synthase, a 2-MDa trimeric protein complex known as 6-deoxyerythronolide B synthase (DEBS)3. This megasynthase is comprised of three unique homodimers assembled from the gene products DEBS1, DEBS2, and DEBS3, which are housed within the erythromycin biosynthetic gene cluster. Each homodimer contains two clusters of catalytically independent enzymatic domains, or modules. Each module, in turn, catalyzes one round of polyketide chain extension and modification (Figure 1). To do so, every chain-extending module includes a ketosynthase (KS), an acyl transferase (AT), and an acyl carrier protein (ACP) domain, in addition to optional enzymes that modify the growing chain such as a ketoreductase (KR), a dehydratase (DH), and/or an enoyl reductase (ER) domain. Polyketide biosynthesis is initiated by the loading didomain (LD), whereas the 6-deoxyerythronolide B product is released by the thioesterase (TE) domain.
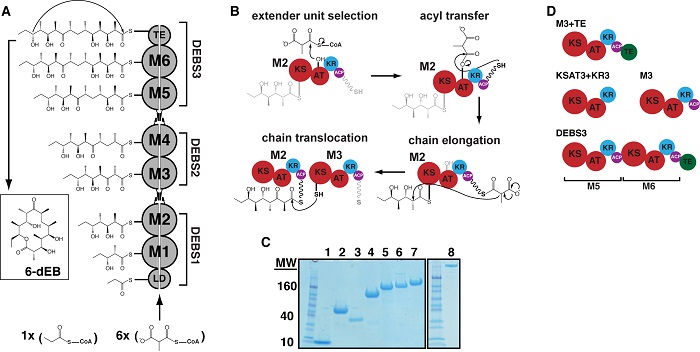
Figure 1. Biosynthesis of 6-deoxyerythronolide B (6dEB) on the DEBS assembly line. (A) DEBS is comprised of six extension modules (M1-6), a loading didomain (LD), and a terminal thioesterase domain (TE). These enzymes are dispersed among three homodimeric polypeptides (DEBS1-3). Successive polypeptides in the assembly line associate through specific docking domain interactions localized near the N- and C-termini. The LD initiates polyketide synthesis with a propionyl-CoA derived primer that is incrementally elaborated as it traverses M1-M6. The TE releases 6dEB via concomitant cyclization. (B) The chain elongation modules are comprised of homologous domains. Acyl transferase (AT) domains transfer methylmalonyl extender units to their acyl carrier protein (ACP) partner domains. The ACP then associates with the ketosynthase (KS) domain from the same module to enable elongation of the polyketide chain. Following elongation, the ACP-bound chain can be modified by auxiliary domains such as the ketoreductase (KR), dehydratase (DH; not shown), and the enoyl reductase (ER; not shown). The fully processed polyketide intermediate is eventually translocated from this ACP to the KS domain of the downstream module. (C) SDS-PAGE analysis of purified proteins prior to size-exclusion chromatography (SEC) and small-angle x-ray scattering (SAXS). Protein samples are as follows: 1) holo-ACP3, 2) KR1, 3) TE, 4) KSAT3, 5) KSAT3+KR3, 6) M3, 7) M3+TE, and 8) DEBS3. (D) Schematic representation of the constructs analyzed in this study using SAXS. All but one construct in the series is derived from M3. By fusing the TE domain onto M3, the M3+TE homodimer is capable of catalyzing multiple turnover in vitro . The bimodular construct DEBS3 is as shown in (A).
Since the discovery of the modular nature of PKS assembly lines4,5, considerable research has focused on engineering PKS chimeras by swapping domains in and out of modules as well as mixing and matching phylogenetically distinct modules to produce new compounds. While this strategy is sometimes effective, the engineered systems are invariably inefficient, underscoring the importance of pursuing a deeper understanding of the relationship between PKS structure and function.
In a recent study, published in the Journal of Molecular Biology, researchers from Stanford University and SSRL used the state-of-the-art capabilities of SSRL’s Beam Line 4-2, which is dedicated to biological small-angle x-ray scattering (SAXS) and diffraction experiments, in order to examine the architecture of DEBS. SAXS is capable of resolving the relative orientations of structurally defined domains within large, flexible protein complexes that resist crystallization, making it an ideal technological platform for probing the structure of DEBS.
In their report, the scientists describe size-exclusion chromatographic separation coupled with small-angle x-ray scattering (SAXS) analyses of a whole module and bimodule from DEBS as well as a set of domains for which high-resolution structures are available. In all cases, the solution state was probed under previously established conditions that ensure each protein is catalytically active. SAXS data are consistent with atomic-resolution structures of DEBS fragments. Therefore, the research team used the available high-resolution structures of DEBS domains to model the architectures of the larger protein assemblies using rigid body refinement. The molecular envelope of DEBS3 (660-kDa homodimer comprising modules 5 and 6) is a thin, elongated ellipsoid, and the results of rigid body modeling suggest that modules 5 and 6 stack collinearly along the 2-fold axis of symmetry (Figure 2).

During polyketide biosynthesis, the ACP covalently and sequentially shuttles the growing polyketide chain to each active site in a module, and ultimately translocates the nascently elongated and modified chain to the next module in the assembly line. Although the resolution accuracy of the SAXS datasets were not high enough to allow the precise modeling of the spatial orientation of the ACP with respect to all of its partner domains, the researchers were able to verify that dramatic conformational distortions of the PKS module and bimodule shapes were not required for the ACP to access its partner enzymes. Using rigid body modeling in CORAL, the scientists simulated domain dynamics along the catalytic cycle of both module 3 and DEBS3 by applying a distance constraint of 20 Å between the appropriate ACP domain and the active site of each catalytic domain in the construct. They observed that the theoretical scattering curve for the resulting structures fit the experimental data comparably to models built without imposing any constraint on the position of the ACP6. Thus, the overall disc-shaped structure of module 3 and the collinear arrangement of modules within DEBS3 appear to be geometrically consistent with catalytically competent enzymes because the ACP domains can be positioned within 20 Å of each active site without dramatically changing the macromolecular architecture. However, the precise spatial positions and protein-protein interactions that each domain samples during catalysis will require higher-resolution insights, which, in turn, will unquestionably enhance the current understanding of assembly line PKS function and the ability to engineer these remarkable megasynthases for the artificial production of natural products.
The modular architecture of PKS assembly lines is a critical feature that facilitates the evolutionary process by allowing bacteria to rapidly mix and match or duplicate sections of PKS assemblages, creating the potential to produce novel antimicrobial analogs. The models of DEBS3, derived from the recent study, suggest that intermodular interactions are minimal, supporting biochemical evidence that the binding affinity between adjacent modules in an operating assembly line is on the order of 1 μM. Similarly, PKS assembly line modules are moderately unrestrained with regard to partnering with other modules, suggesting that low affinity maybe accompanied by low specificity, as evidenced by the ability of biological engineers to mix and match modules from divergent phylogenetic backgrounds with reasonable success and very minimal engineered protein-protein interactions.
Taken together, a collinear arrangement of modules with minimal protein-protein interactions may facilitate evolution of new assembly lines by allowing whole module duplication events7–9 within a functioning PKS assembly with a relatively low impact on activity. Alternatively, homologous recombination occurring at intermodular junctions between distinct assembly lines is an attractive model for driving PKS assembly line diversity10–12. In either case, a malleable architecture would allow competing bacteria to rapidly produce novel antibiotics and signaling compounds from rather limited genetic resources. The arrangement of domains within a whole module and bimodule, reported in the recent study, represents a critical step forward in the understanding of PKS structural biology as this work sets the stage for a detailed investigation of protein-protein interactions that facilitate intermodular interactions. The technological capabilities provided by SSRL’s Beam Line 4-2 allowed the collection of extremely high-quality data, leading to critical structural insights into the large and flexible DEBS proteins.
- A. L. Demain, "The Business of Biotechnology", Ind. Biotechnol. 3, 269 (2007).
- W. Minas, Microbial Processes and Products. 65–90 (2005).
- C. Khosla, Y. Tang, A. Y. Chen, N. A. Schnarr and D. E. Cane, "Structure and Mechanism of the 6-deoxyerythronolide B Synthase", Annu. Rev. Biochem. 76, 195 (2007).
- S. Donadio, M. J. Staver, J. B. McAlpine, S. J. Swanson and L. Katz, "Modular Organization of Genes Required for Complex Polyketide Biosynthesis", Science 252, 675 (1991).
- J. Cortes, S. F. Haydock, G. A. Roberts, D. J. Bevitt and P. F. Leadlay, "An Unusually Large Multifunctional Polypeptide in the Erythromycin-producing Polyketide Synthase of Saccharopolyspora erythraea", Nature 348, 176 (1990).
- A. L. Edwards, T. Matsui, T. Weiss and C. Khosla, "Architectures of Whole-Module and Bimodular Proteins from the 6-Deoxyerythronolide B Synthase", J. Mol. Biol. 426, 2229 (2014).
- M. A. Fischbach, C. T. Walsh and J. Clardy, "The Evolution of Gene Collectives: How Natural Selection Drives Chemical Innovation. Proc. Natl. Acad. Sci. USA 105, 4601 (2008).
- H. Jenke-Kodama and E. Dittmann, "Evolution of Metabolic Diversity: Insights from Microbial Polyketide Synthases", Phytochemistry 70, 1858 (2009).
- H. Jenke-Kodama, H., Börner, T. and E. Dittmann, "Natural Biocombinatorics in the Polyketide Synthase Genes of the Actinobacterium Streptomyces avermitilis", PLoS Comput. Biol. 2, 1210 (2006).
- J. R. Doroghazi, and D. H. Buckley, "Widespread Homologous Recombination within and between Streptomyces species", ISME J. 4, 1136 (2010).
- A. Starcevic et al., "A Novel Docking Domain Interface Model Predicting Recombination between Homoeologous Modular Biosynthetic Gene Clusters", J. Ind. Microbiol. Biotechnol. 38, 1295 (2011).
- J. Zucko, P. F. Long, D. Hranueli and J. Cullum, "Horizontal Gene Transfer and Gene Conversion Drive Evolution of Modular Polyketide Synthases", J. Ind. Microbiol. Biotechnol. 39, 1541 (2012).
A. L. Edwards, T. Matsui, T. M. Weiss and C. Khosla, "Architectures of Whole-Module and Bimodular Proteins from the 6-Deoxyerythronolide B Synthase", J. Mol. Biol. 426, 2229 (2014) doi: 10.1016/j.jmb.2014.03.015
