 Christopher C. Fuller,1 John R. Bargar,2 and James A
Davis1
Christopher C. Fuller,1 John R. Bargar,2 and James A
Davis1
1U.S. Geological Survey, Water Resources Division, Menlo Park, CA
2Stanford Synchrotron Radiation Laboratory, SLAC, Stanford
University, Stanford, CA
Clean up of contaminated aquifers is a difficult and expensive problem because of the inaccessibility of the subsurface and the volume of soil and ground water requiring treatment. Established technologies such as pump-and-treat and soil excavation are ineffective in most large contamination scenarios because they treat only a small fraction of the contamination or are prohibitively expensive (National Research Council, 1999). Permeable Reactive Barriers (PRBs) are a relatively new technology that offer promise to overcome these obstacles. PRBs are trenches (figure 1) or fence-like arrays of non-pumping wells emplaced in the subsurface at depths of up to 150 feet to intercept the flow of contaminated ground water (Freethey et al., 2002). Fill materials contained within the PRBs react with dissolved contaminants to degrade or sequester them. Thus, in essence, PRBs act as large in-situ filters to clean ground water. PRB technologies offer lower operating costs, are highly energy efficient, and require no surface facilities or ground water pumping/recharge (Freethey et al., 2002; Morrison and Spangler, 1992; Shoemaker et al., 1995). Two commonly proposed PRB contaminant-removal mechanisms are: (a) precipitation reactions in which metal contaminants are sequestered within freshly formed mineral phases, and (b) oxidative degradation of contaminants by particulate iron metal. In order for PRBs to be cost-effective they should be effective for an economically viable period (or be replenishable). They should not be susceptible to clogging or rapid passivation by reaction products.

 | |
| Figure 1. Top: schematic diagram of a permeable reactive barrier. Bottom: installation of the apatite PRB at Fry Canyon, Utah. |
The shallow alluvial aquifer at Fry Canyon, Utah, is contaminated with up to 17 mg/L uranium leached from processed tailings at an ore upgrader processing plant that was operated in the 1950s and 60s. A partnership was formed in June of 1996 between the DOE (Grand Junction Office), US EPA, Interior Department, Geological Survey (USGS), Bureau of Land Management (BLM), and the Utah Department of Environmental Quality (UDEQ) for the purposes of demonstrating PRB in-situ treatment technologies for abating the ground water uranium contamination.
Commercial apatite (Ca5(PO4)3OH), was investigated for use at Fry Canyon based on its reactivity with uranium. Apatite is soluble in ground water and slowly releases phosphate, which can react with dissolved hexavalent uranium (U(VI)) to form the mineral autunite (Ca(UO2)2(PO4)2·10H2O)), which is pictured in the highlight announcement on the main page. The relatively low solubility of autunite has led some to propose that it would be an ideal inert host for in-situ sequestration and immobilization of U(VI). This general chemical strategy (phosphate mineral formation) has previously been shown to have a high capacity for attenuating lead- and cadmium contamination (Ma et al., 1993; Valsami-Jones et al., 1998).
Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy and Synchrotron-based X-Ray Diffraction (SR-XRD) measurements were performed at
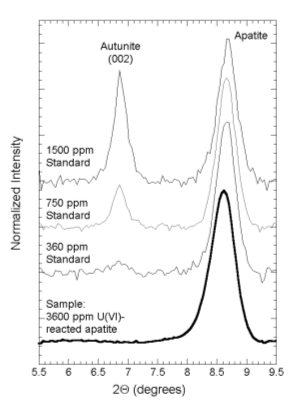 | |
| Figure 2. XRD region scans of uranium-reacted apatite contain no evidence for presence of autunite. Detection limit for autunite is 360 ppm, which is a 10-fold lower concentration than in the sample. (from Fuller et al., 2002) |
It follows from these measurements that U(VI) probably occurs in the PRBs as molecular complexes adsorbed onto the surfaces of apatite. EXAFS analyses are consistent with this conclusion. EXAFS spectra suggest that uranium occurs as the uranyl cation (U(VI)O22+) bonded to apatite surfaces and complexed by carbonate groups (Fuller et al., 2003). The uranium-phosphorous distance is about 3.55 Ĺ, suggesting that U(VI) coordinates to surface phosphate groups in a monodentate geometry (figure 3). These results were obtained from laboratory samples and samples extracted from the Fry Canyon barrier after 18 months of operation. Thus, uranium sequestration in the barriers is believed to occur primarily via adsorption of U(VI)-carbonate complexes onto the surfaces of apatite PRB material. These results have the important implication that permeability will remain high in the apatite PRB. In contrast, clogging in zero-valent-iron-based PRBs from precipitation of corrosion products can seriously impact the longevity of the PRB. However, the barrier will require monitoring to insure that re-release of sequestered U(VI) does not occur due to changes in ground-water chemistry or if the U(VI) sorption equilibrium is reached.
 |
| Figure 3. LIII-edge EXAFS and Fourier Transforms (FTs) of autunite and uranium-reacted apatite (3600 ppm sorbed U(VI)). Solid lines are data. Dotted lines are fits. Structural drawings: Blue = U(VI) (axial oxygens above and below plane of page are omitted), Red = Oxygen, Green = Phosphorous. The differences in the local structure around uranium (right hand side) give rise to the differences observed in the EXAFS and FTs. |
References
- Freethey G. F., Naftz D. L., Rowland R. C., and Davis J. A. (2002) Deep Aquifer Remediation Tools: Theory, Design, and Performance Modeling. In Handbook of Groundwater Remediation Using Permeable Reactive Barriers (ed. D. L. Naftz, S. J. Morrison, J. A. Davis, and C. C. Fuller), pp. 133-163. Academic Press.
- Fuller C. C., Bargar J. R., and Davis J. A. (2003) Molecular-scale characterization of uranium sorption by bone apatite materials for a permeable reactive barrier demonstration. Environ. Sci. Technol. 37, 4642-4649.
- Fuller C. C., Bargar J. R., Davis J. A., and Piana M. J. (2002) Mechanisms of uranium interactions with hydroxyapatite: implications for ground water remediation. Environ. Sci. Technol. 36, 158-165.
- Ma Q. Y., Traina S. J., Logan T. J., and Ryan J. A. (1993) In situ lead immobilization by apatite. Environ. Sci. Technol. 27, 1803-1810.
- Morrison S. J. and Spangler R. R. (1992) Chemical barriers for controlling groundwater contamination: Environ. Prog. 12, 175-181.
- National Research Council. (1999) Groundwater and Soil Cleanup. National Academy Press.
- Shoemaker S. H., Greiner J. F., and Gillham R. W. (1995) Permeable Reactive Barriers. In Assessment of Barrier Containment Technologies (ed. R. R. Rumer and J. K. Mitchell), pp. 301-353. National Technical Information Service.
- Valsami-Jones E., Ragnarsdottir K. V., Putnis A., Bosbach D., Kemp A. J., and Cressey G. (1998) The dissolution of apatite in the presence of aqueous metal cations at pH 2-7. Chem. Geol. 151, 215-233.
SSRL Highlights Archive