
 Hongjun Liang,* Daniel Harries,+ and Gerard C. L.
Wong*
Hongjun Liang,* Daniel Harries,+ and Gerard C. L.
Wong*
*Department of Materials Science & Engineering, Department of Physics, Department of Bioengineering, University of Illinois at Urbana-Champaign, IL 61801, USA
+Laboratory of Physical and Structural Biology, National Institute of Child Health and Human Development, NIH, Bethesda, MD 20892, USA
Gene therapy using either viral or synthetic vectors is currently one of the
most promising strategies for developing cures for many hereditary and acquired
diseases. Protocols have been approved for cancer, hemophilia, cystic fibrosis,
neuromuscular disorders, and others. Although synthetic nonviral systems such
as cationic liposomes generally transfect less efficiently than viruses, they
have a number of advantages such as high DNA packaging capacity and low
immunogenicity. Since their introduction in 1987, cationic lipid-DNA complexes
(CL-DNA) have emerged as one of the major non-viral DNA delivery platforms.
CL-DNA complexes have been used in gene therapy for a broad range of cell types
as well as delivery systems for cancer vaccines.
Anionic lipids (AL) occur naturally in eukaryotic cell membranes, and DNA
delivery systems based on anionic lipids have recently been examined as an
alternative to cationic lipids due to their low cytotoxicity. Anionic lipids
can be complexed with anionic DNA via interaction with multivalent cations such
as Ca2+, and have been shown to transfer oligonucleotides into
hippocampal neurons and bacterial cells. An outstanding problem of this
approach is the inefficient association between the anionic lipids and DNA
molecules, which is attributed to their like-charge electrostatic repulsion.
Rational design of AL-DNA vectors requires a coherent understanding of their
structures and interactions. A useful starting point is the physics governing
the analogous CL-DNA complexes that has emerged in the last few years. The
addition of DNA to cationic lipid mixtures induces a topological transition
from liposomes into condensed multi-lamellar self-assemblies, where parallel
DNA chains are confined between lipid sheets. By lowering the membrane's
bending rigidity or by changing its spontaneous curvature, an inverted
hexagonal phase with an enhanced tendency for membrane fusion can be formed, in
which DNA chains coated by lipid monolayers are packed into a 2D columnar
hexagonal array. In these self-assembled complexes, the cationic lipid head
groups neutralize the phosphate groups on the DNA chains, effectively releasing
the counterions previously bound electrostatically to lipids and DNA, thus
gaining translational entropy in the bulk. The pioneering studies have shown
that physical parameters, such as self-assembled nanostructure and membrane
charge density, are crucial elements in transfection efficiency.
In this work, the structure and interactions of AL-DNA complexes in the
presence of different divalent cations have been systematically investigated
using confocal microscopy and synchrotron small angle X-ray scattering (SAXS)
on Beam Lines 4-2 at SSRL and 34ID at the Advanced Photon Source. AL-DNA
complexes are governed by more complex interactions than that for their CL-DNA
analogues. While cationic membranes are attracted to DNA mainly via entropic
forces due to counterion release, anionic membranes require multivalent cations
to mediate attractions to anionic DNA through direct electrostatic "bridging"
interactions. Further, the addition of multivalent ions can mediate strong
attractions between different combinations of membranes and DNA and induce the
formation of not just condensed DNA-membrane complexes, but also potentially
condensed membrane complexes and condensed DNA complexes, both of which have no
analog in CL-DNA systems. Finally, divalent cations can also coordinate
non-electrostatically with lipid molecules and modify membrane structure.
We find from the SAXS measurements that at low membrane charge densities,
AL-DNA complexes self-assemble into a lamellar structure, with alternating
layers of like-charged DNA and anionic membranes bound together with divalent
cations (A). As the membrane charge density is increased, we find a new phase
with no analog in CL-DNA systems: DNA is systematically expelled from the
complex, and the divalent ions mediate attractions between anionic membrane
sheets to form a lipid lamellar stack (B). Divalent ions differ in their
tendency to coordinate non-electrostatically with lipids. Zn2+ ions
are known to have strong non-electrostatic interactions with lipids, involving
significant dehydration of the lipid headgroups, while others such as
Mg2+ have a much smaller effect. The SAXS data show that as the
global Zn2+ concentration is increased, both lamellar phases are
destabilized. The system instead forms an inverted hexagonal phase, comprised
of a hexagonal array of divalent cation coated DNA strands wrapped in turn by
anionic membrane monolayers (C). While Zn2+ is known to adhere to
both lipids and DNA, we suggest that the change in AL-DNA structure is
primarily due to a cation-induced change in the membrane spontaneous curvature
c0. Using simple theoretical considerations, we show that
the expected cross-over between the lamellar phase and hexagonal phase occurs
at a critical c0 close to the experimentally observed values.
Primary Citation
Liang, H.J., Harries, D. & Wong, G.C.L. Proc. Natl. Acad. Sci. USA
102, 11173-11178. (2005)
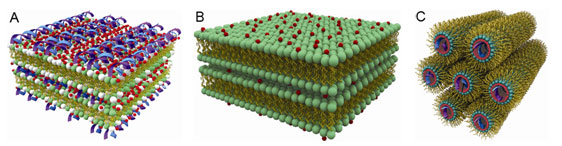
Schematic pictures of (A) Condensed DNA-ion-membrane lamellar structure with
alternating layers of DNA and anionic membranes 'glued' together by divalent
cations; (B) Condensed ion-membrane lamellar structure in which charged
membranes stacks are held together by divalent cations; and (C) 2-D inverted
hexagonal structure in which hexagonal arrays of divalent cations coated DNA
strands wrapped in the anionic membrane monolayer tubes.
| SSRL is supported by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. |